Overview
Aspect
merlin main window is divided in several sections. Firstly, the "Operations Menu" includes workspace configuration and all the major model reconstruction related operations. The ‘Workspace Dashboard’ contains the opened workspaces, as well as the composing dashboards "model", "annotation" and "validation". Upon "opening" a selected dashboard, several "boards" become available.
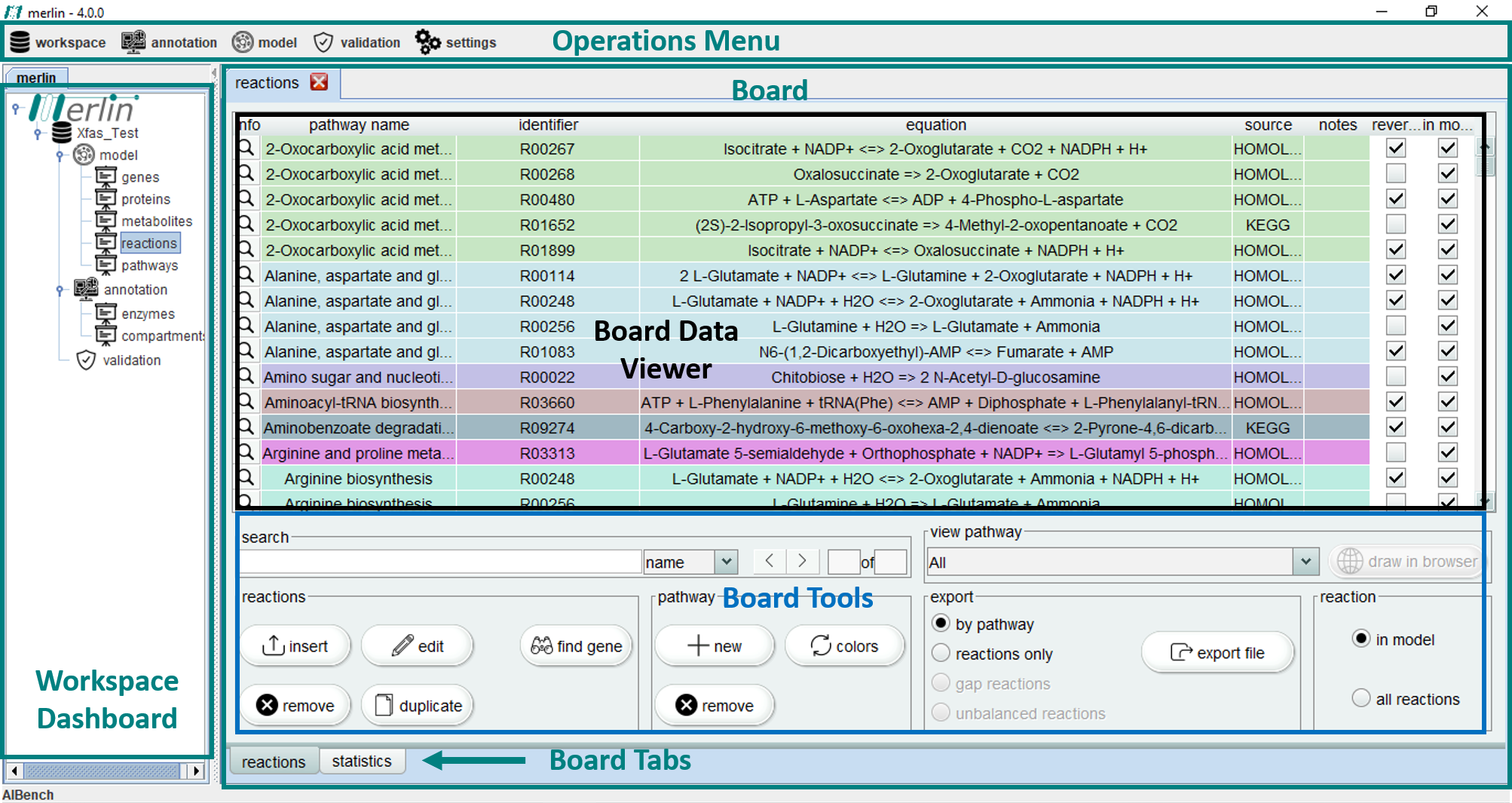
Finally, the major part of merlin window is filled by selecting the intended board, in the workspace dashboard section. This board is composed by different sections, including:
-
Board Data Viewer - Display of useful data of the selected board;
-
Board Tools - operations related to the current displayed data;
-
Board Tabs - These can be used to cycle between the different tabs of a board.
Before starting
Recommendations
Before you start we suggest the following optional procedures to enhance the reconstruction of Genome-Scale Metabolic Models using merlin:
-
BLAST+ - Consider installing NCBI's standalone BLAST+ tool. This tool can be used for hastening the GPRs procedure. Follow the instructions: Windows; Unix; Others;
-
Memory - Consider increasing the maximum heap memory size (RAM) for large genome sequences or multiple reconstructions. See Increase maximum heap memory size for more details.
Open merlin
Unzip the merlin folder that you have just downloaded and double-click on the merlin.bat file.
Database settings
If the user wants to use merlin’s default database settings, the following instructions can be skipped. Otherwise, to select the database type, the user must open the section “settings” in the menu and then select “database”. Here, the database type is the first option, where the option H2 is the local database and is selected by default. Alternatively, merlin also supports connections to local or remote MySQL databases when the following fields are set: username, password, host, and port. When all parameters are set, merlin should be able to successfully establish a connection with the user’s database server.

Workspace
Create a workspace
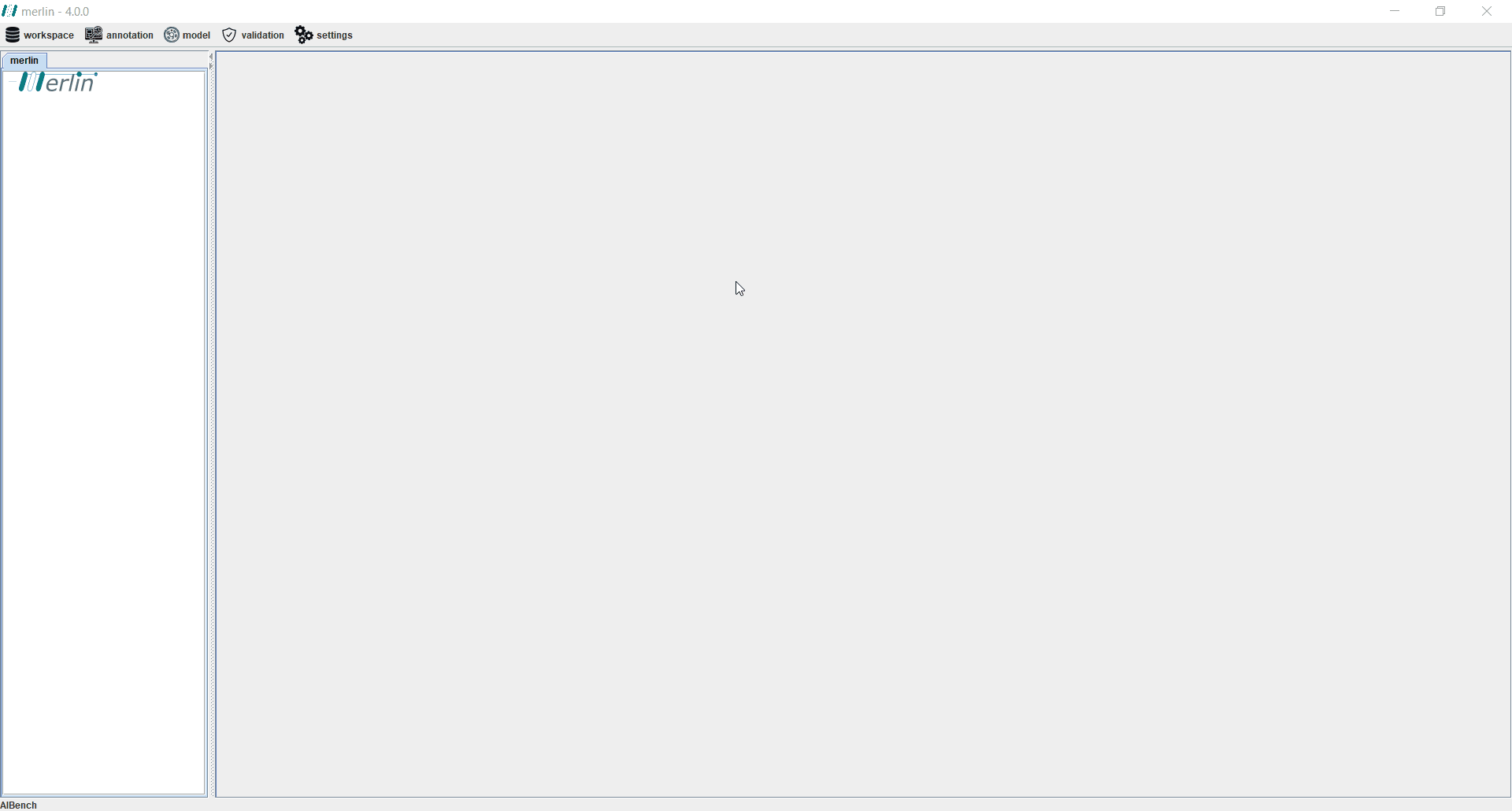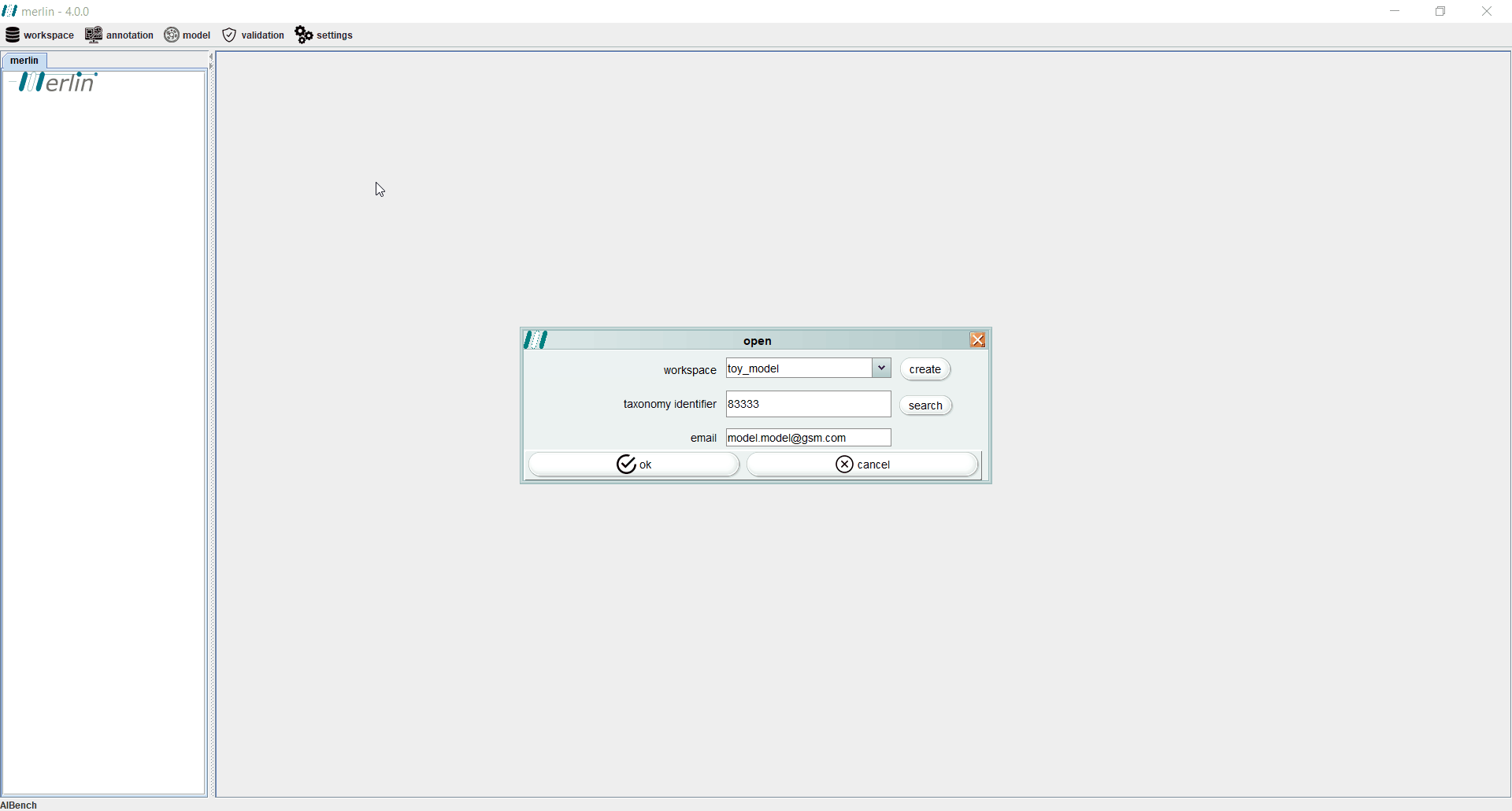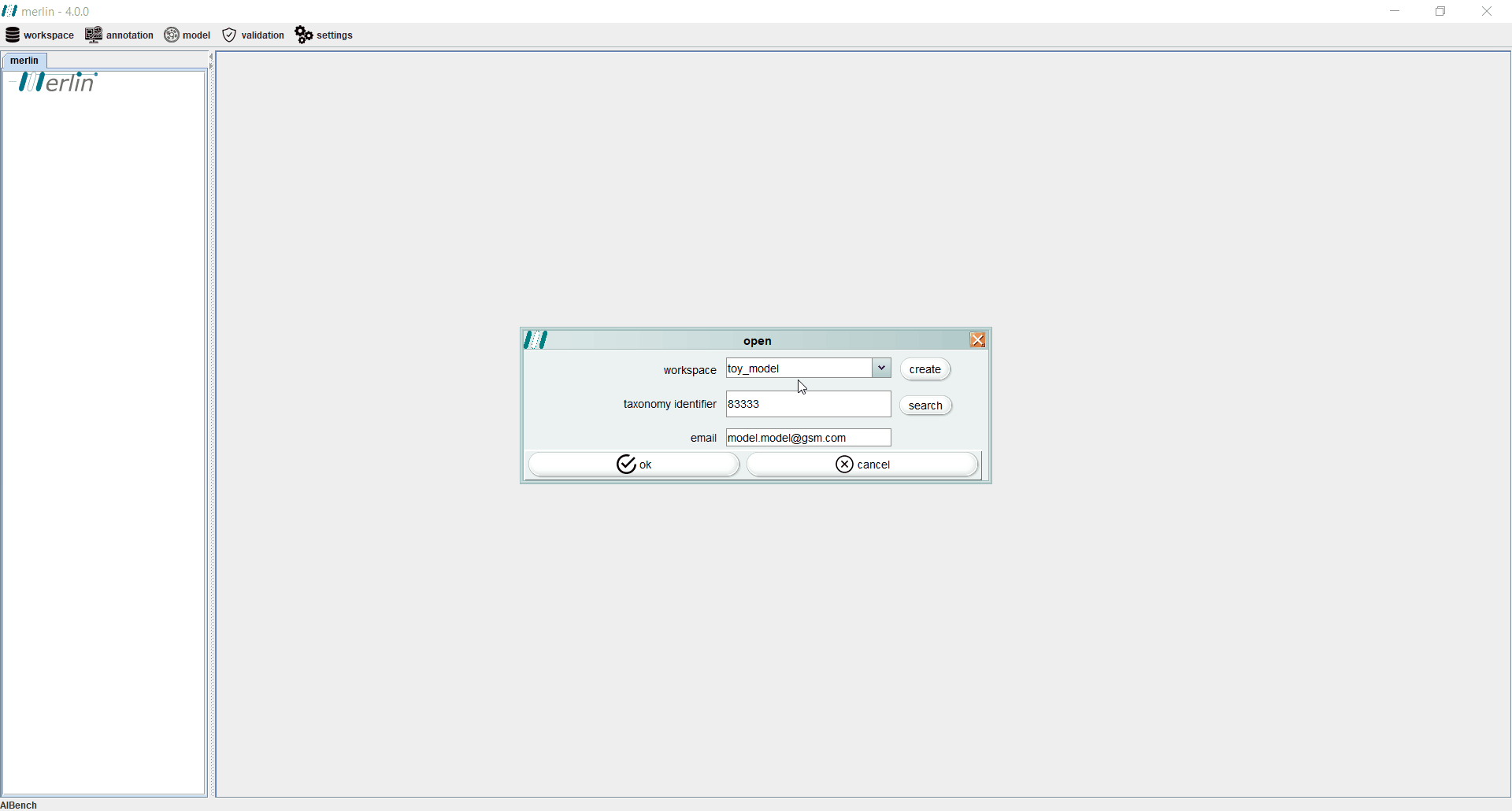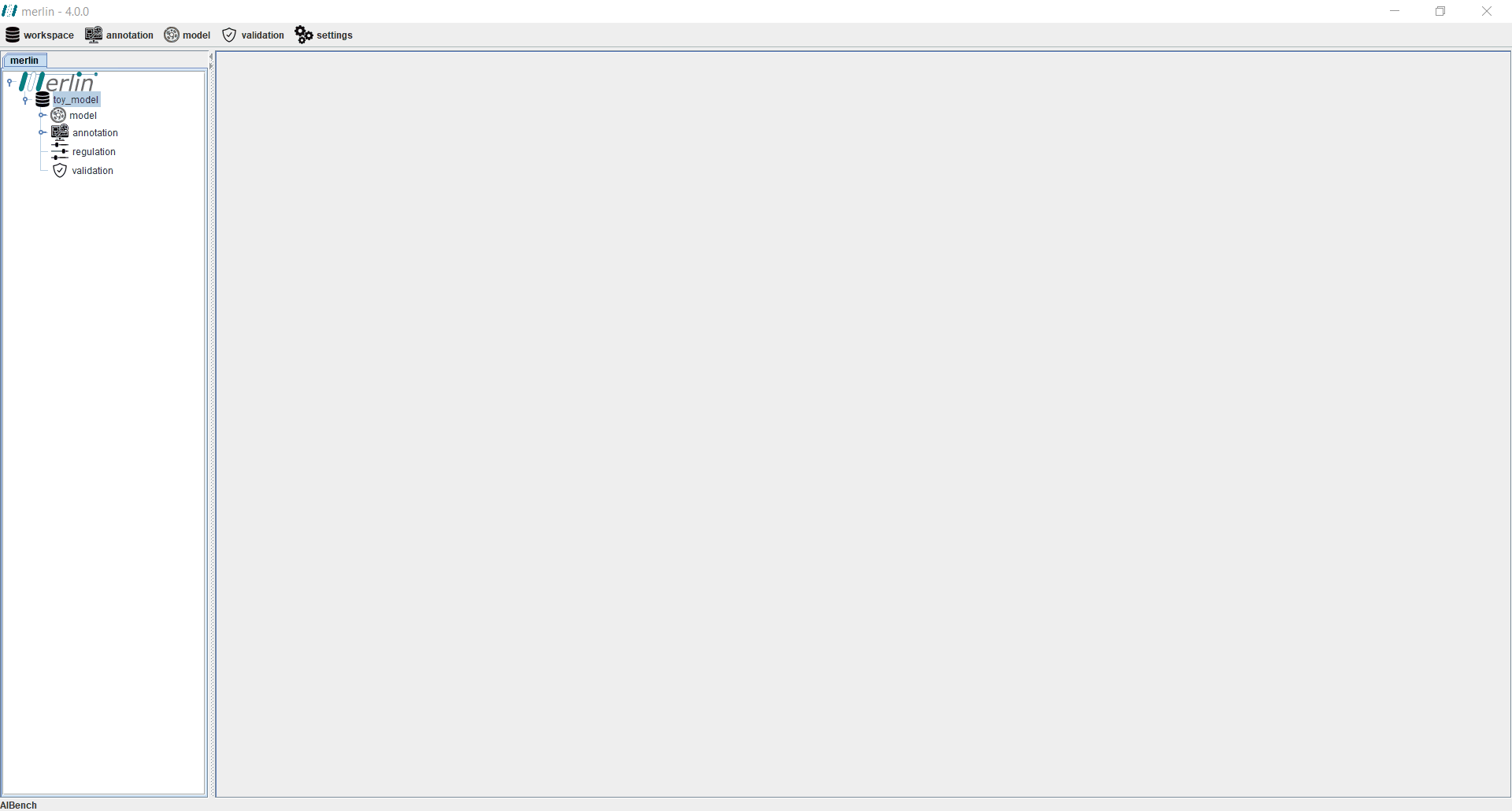
To create a new workspace, access the “workspace” section and select the “open” option. When a workspace is being created for the first time, merlin will prompt users to automatically create a workspace. To create a workspace at any given time, select the “create” option and enter the new workspace name (duplicate names are not possible and a warning will be prompted).
After creating a workspace it is mandatory to enter the NCBI Taxonomy Identifier of the organism in study. When available, all NCBI files associated with a given assembly record may be automatically download , by selecting the “search” option. The final step towards creating a new workspace consists in entering the user email. This information will be stored in the user’s local machine, as it is mandatory to remotely access certain tools, such as UniProt or BLAST.

Load a workspace
To load an existing workspace into merlin access the “workspace” section and select “open”. Then, select the desired workspace from a list of available options. The taxonomy identifier and personal email are automatically reloaded.
Import a workspace
merlin allows importing existing workspaces, regardless of origin. To import a workspace access the “workspace” section and hover over the “import” option, and select the “workspace” option. A new panel in which users can browse the local drive to select the merlin workspace backup (“.mer”), will emerge. Additionally, it is possible to redefine a new name for the imported workspace. If the user decides not to provide a new name, the imported workspace will inherit its original name, unless a workspace with the same name is already present in which case a warning will be shown.

Export a workspace
merlin provides an option to export a backup of a given workspace, which can be used for personal backup or sharing with other users. To export a workspace access the “workspace” section, hover over the “export” option and select “workspace”. A new panel in which users can select the workspace to export, and where to in the local drive, will emerge. Workspaces are exported in the “.mer” format and are automatically labeled based on the workspace name and time/date of export.

Clean a workspace
“Cleaning” a workspace consists in deleting specific parts of the database associated with the workspace. To clean a workspace access the “workspace” section and select the “clean” option. A panel will emerge asking users to select the target workspace. Additionally, users can select which section should be wiped, namely:
whole database of the workspace;
model data only;
enzymes annotation;
compartments annotation.
Moreover, advanced options are available, such as to maintain/delete gene sequences and associated data, automatically reloading KEGG’s metabolic data, etc.

Delete a workspace
Deleting a workspace is a permanent operation which will delete the associated database. To delete a workspace access the “workspace” section and select the “delete” option. A panel will emerge asking for the target workspace to delete. Notice that the workspace must be opened to be deleted.

Clone a workspace
merlin easily allows users to make a copy of available workspaces efficiently without requiring additional export and import operations. To clone a workspace access the “workspace” section and click the “clone workspace” section. A new panel will emerge asking users to select the workspace to clone and the name of the generated copy. If a user enters a name for a copy which matches an already existing workspace, the operation will not continue. If the user does intend to replace an existing workspace with a copy of another workspace, this restriction can be bypassed by selecting the “force database creation” option.

Compatibility (import workspace from merlin v3 to merlin v4)
To import a workspace from merlin 3 to merlin 4, the user must open the operation located at “workspace” -> “compatibility” -> “import workspace from merlin 3”. A window should open where the user must indicate the name of the workspace to be generated in merlin 4. Note that this name should not already exist in the database, otherwise merlin will show an error. However, the user can force merlin to override this database by selecting the option to “force database creation”. The second field is the name of the workspace to be imported from merlin 3. The last field is to indicate the path to merlin 3 home folder.
Genomic data
Genome sequence files
Before starting the genome annotation stage it is necessary to provide the genome data to merlin. The genome of the organism in study is the cornerstone for most steps required for the reconstruction of the metabolic model. Genome data may be uploaded into merlin with files or by accessing NCBI remotely. Additionally, it is also possible to automate the retrieval of genome files upon creating a new workspace. After entering the NCBI Taxonomy Identifier of the organism in study, and clicking the “search” option, it is possible to download the genome data from GenBank or RefSeq (and automatically upload it into merlin).
Import files
Besides importing automatically from NCBI, merlin allows loading local genome files. To perform this operation, the “workspace” section should be accessed, then hovering over the “import” option and selecting the “genome” option. A new panel will emerge with additional settings. The “search locus tags” option, if activated, triggers an operation to associate each given gene (in the genome files) to a locus tag (when available). merlin is compatible with multiple genome file formats, such as “.faa”, “.fna” and “.gbff”. Upon selecting the genome file to be imported, the final step consists in selecting the desired workspace for the data to be loaded into.

Download NCBI files
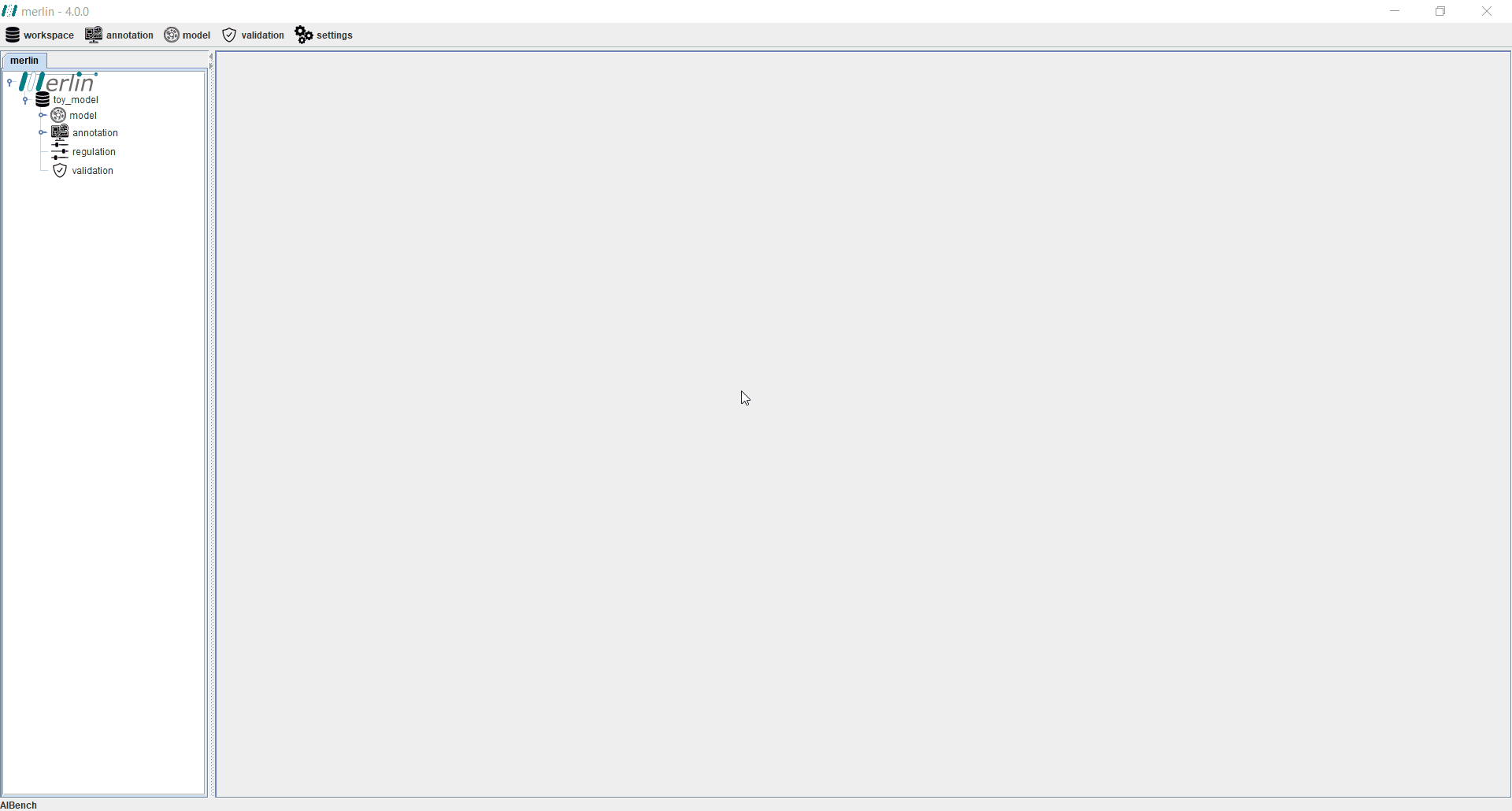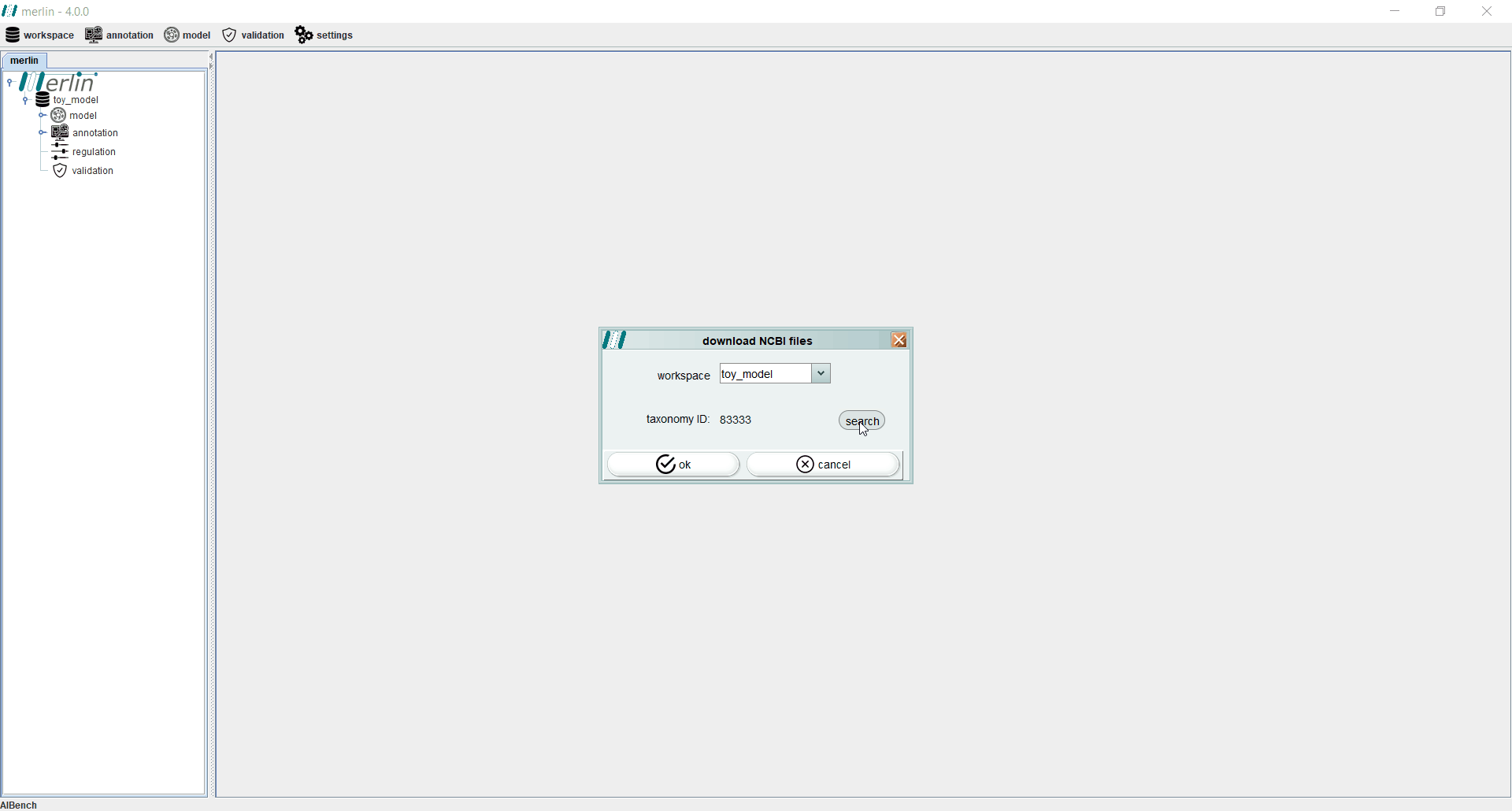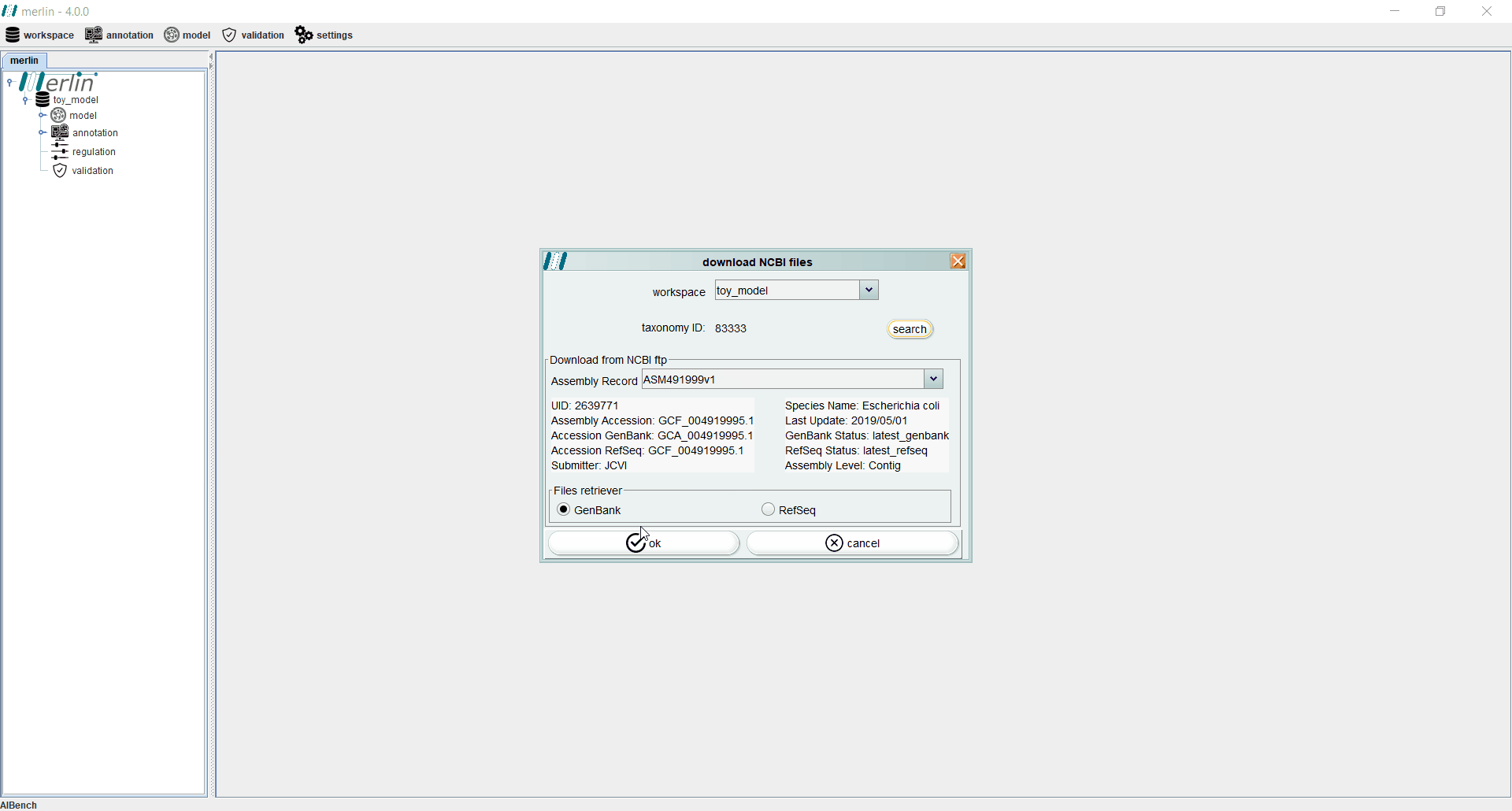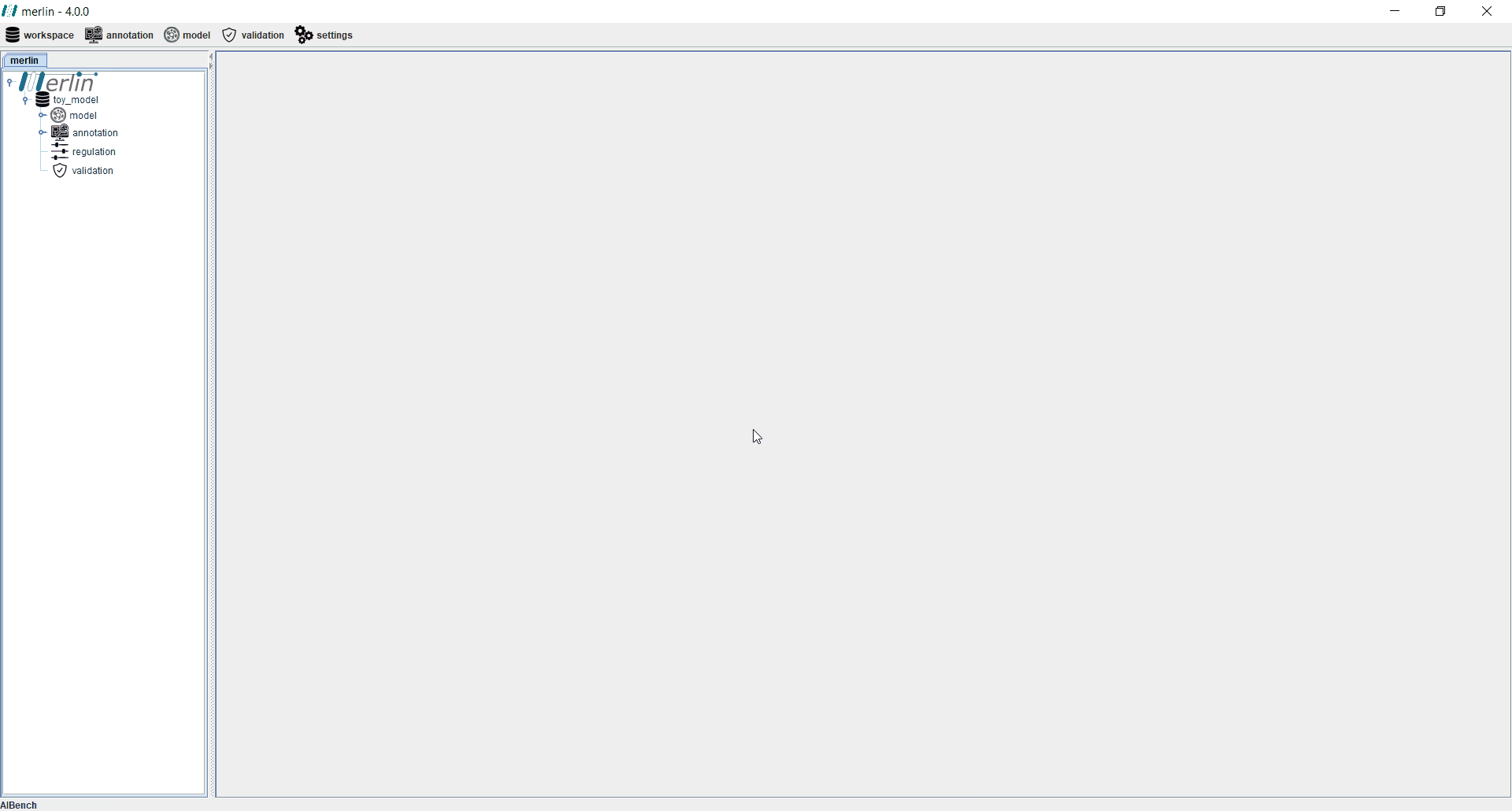
Even if the genome files were not downloaded from NCBI during the creation of a new workspace, users may perform this operation separately by accessing the “workspace” section and selecting the “download NCBI files” option. If necessary, the “search” can be used to find a specific assembly record from GenBank or RefSeq.
Export files
The genome data available in merlin may be exported by accessing the “workspace” section, hovering over the “export” option, and selecting the “genome files” option. A new panel will emerge allowing to select the genome file to be exported and the output directory.

Metabolic data
Load KEGG metabolic data
Obtaining metabolic information is mandatory for a GSM model reconstruction. Therefore, merlin contains a simple operation which retrieves this information from KEGG. This operation will load the metabolic information including compounds, enzymes, reactions and pathways into merlin’s internal database. Currently, merlin only supports KEGG. However, in the near future, tools to retrieve data from BiGG and ModelSEED databases will be available (see ongoing work for further information). This operation automatically integrates all retrieved spontaneous reactions into the model, which will be complemented upon integrating the enzymatic and transporters annotation.

Genome annotation
Creating a genome annotation
One of the most relevant and differentiating features in merlin is that it allows to perform the genome functional annotation of the organism of interest. This coincides with the first step towards the reconstruction of your GSM model. There are several options for performing the genome functional annotation with merlin involving homology or domain searches to the genome sequence of the organism of interest. Nevertheless, external genome functional annotations might be uploaded into merlin (see Load genome functional annotation section).
The purpose of the enzymes annotation, is to assign metabolic functions, namely the gene products and enzyme commission (EC) numbers, to all coding sequence (CDS) encoding enzymes in the genome.
Homology search
In merlin, the enzymes functional annotation procedure is supported by semi-automatic similarity searches to remote databases, such as UniProtKB or UniProtKB/SwissProt, to find homologous protein sequences. Performing an homology search in merlin requires having imported the genome files (see Genome Files section).
Using BLAST
The Basic Local Alignment Search Tool (BLAST) is the preferred similarity search tool for performing the enzymes functional annotation in merlin, which uses the remote web-service from EBI.
merlin allows changing all parameters such as:
-
expected value (e-value): Hint: the lower the E-value, the higher the similarity score between two sequences;
-
BLAST database:
-
UniProtKB - reviewed and unreviewed annotation records from UniProtKB and SwissProt;
-
UniProtKB/SwissProt - reviewed records from SwissProt;
-
UniProtKB/TrEMBL - unreviewed annotation records from UniProtKB;
-
Additionally, merlin supports other UniProtKB-based databases
-
the type of the BLAST tool
-
the number of BLAST results
-
selecting the substitution matrix manually or automatically
merlin will perform genome-wide sequence alignments automatically and display the results in a compact layout, the enzymes annotation board (see Enzymes board). This process may take several hours depending on the web-server availability, internet connection and genome size. merlin allow users to annotate genomes, using different BLAST databases for genes without hits in the previous run. That it, after a BLAST search, genes without similarity results can be annotated with different databases by assessing the same operation and selecting a different database.
Load BLAST report
On the other hand, BLAST reports obtained with the NCBI or EBI web/services may be directly loaded into merlin. This process involves providing the path in the computer for the returned output files, the source and type.
merlin will parse the contents within each BLAST report and load the results into a simplified layout called enzymes board (see Enzymes board).
Domain search
Using InterProScan
The InterProScan functional annotation is an optional step, which can provide additional information regarding each gene functional annotation. This tool will provide a functional analysis for each protein sequence by classifying them into families and predicting the presence of domains and important sites. InterProScan is a time-consuming tool, thus the user must provide both lower and upper thresholds to limit the number of annotated records. The results of your InterProScan functional annotation can be checked by clicking the magnifying glass (coloured with a purple background) of each gene entry in the enzymes board (see Enzymes board).
Load genome annotation
Although it is highly recommended that you perform your own genome functional annotation to assess with high confidence the metabolic capabilities in the genome of the organism of interest, merlin allows you to skip this step. Instead, one can load external genome functional annotations from KEGG or standard format files, such as GFF3 and GenBank.
KEGG annotation
The user must provide the organism identifier (also known as organism code) in KEGG, to load a genome functional annotation from KEGG. For example, eco for Escherichia coli K-12 MG1655. merlin will load automatically the enzymes functional annotation available in KEGG for the selected organism and integrate it into the model dashboard together with the previously loaded metabolic data (see Reactions board section).
GFF3 annotation
The user must provide the file path and source, to load a genome functional annotation from an external GFF3 file. Additionally, the EC option checkbox should be selected to allow the integration of metabolic capabilities encoded in the genome (identified by the EC numbers). merlin will load the enzymes functional annotation available in the GFF3 file and integrate it into the model dashboard together with the previously loaded metabolic data (see Reactions board section).
GenBank annotation
The user can load the genome functional annotation available in the GenBank file uploaded in the Genome files section. Alternatively, to load a genome functional annotation from an external GenBank file, the user must provide the path for the respective file. merlin will load the enzymes functional annotation available in the GenBank file and integrate it into the model dashboard together with the previously loaded metabolic data (see Reactions board section).
Pipelines for genome annotation
merlin allows to perform the genome functional annotation following through methodologies. Nevertheless, the genome functional annotation can be manually performed in the enzymes board (see Enzymes board section).
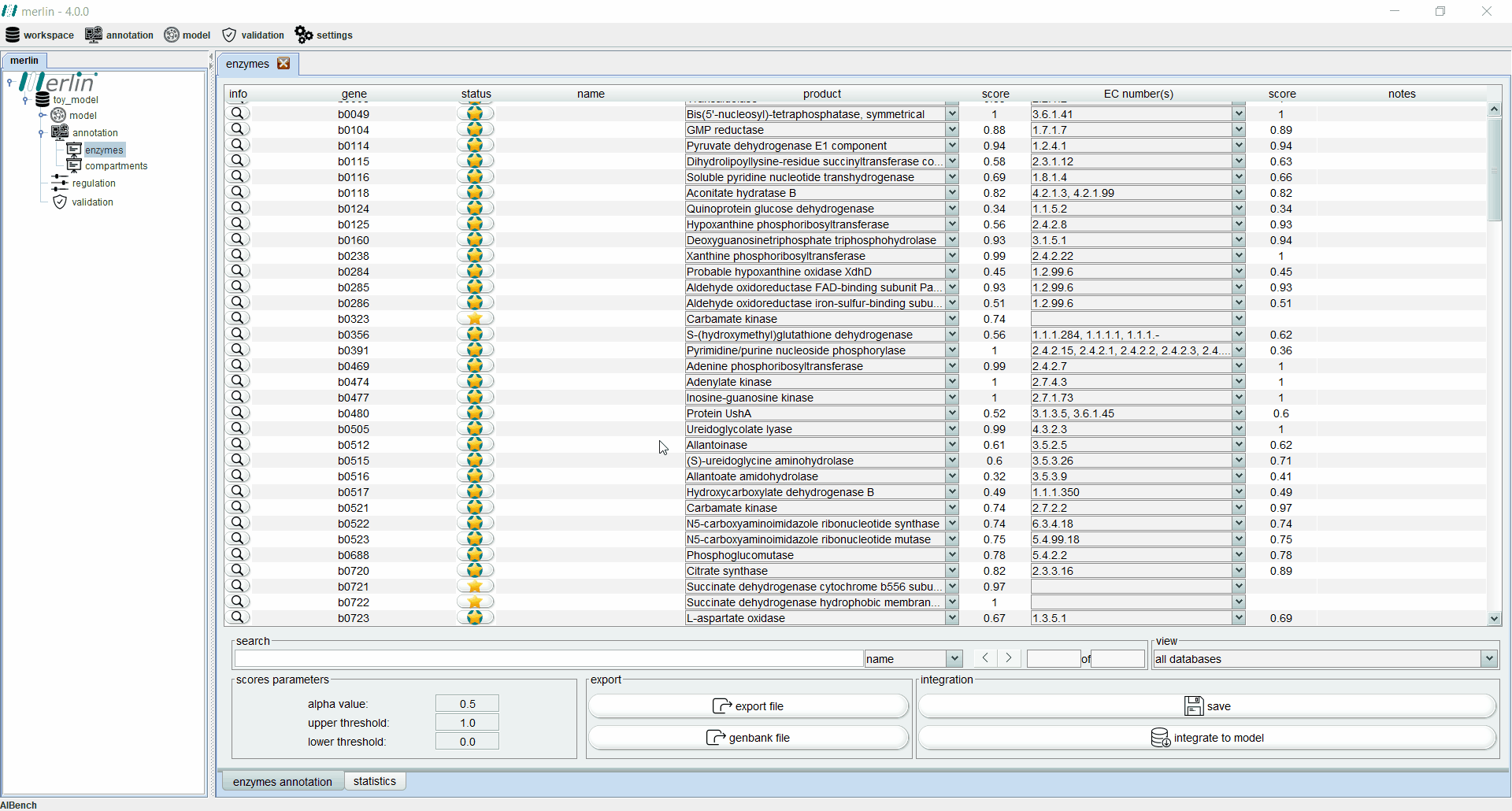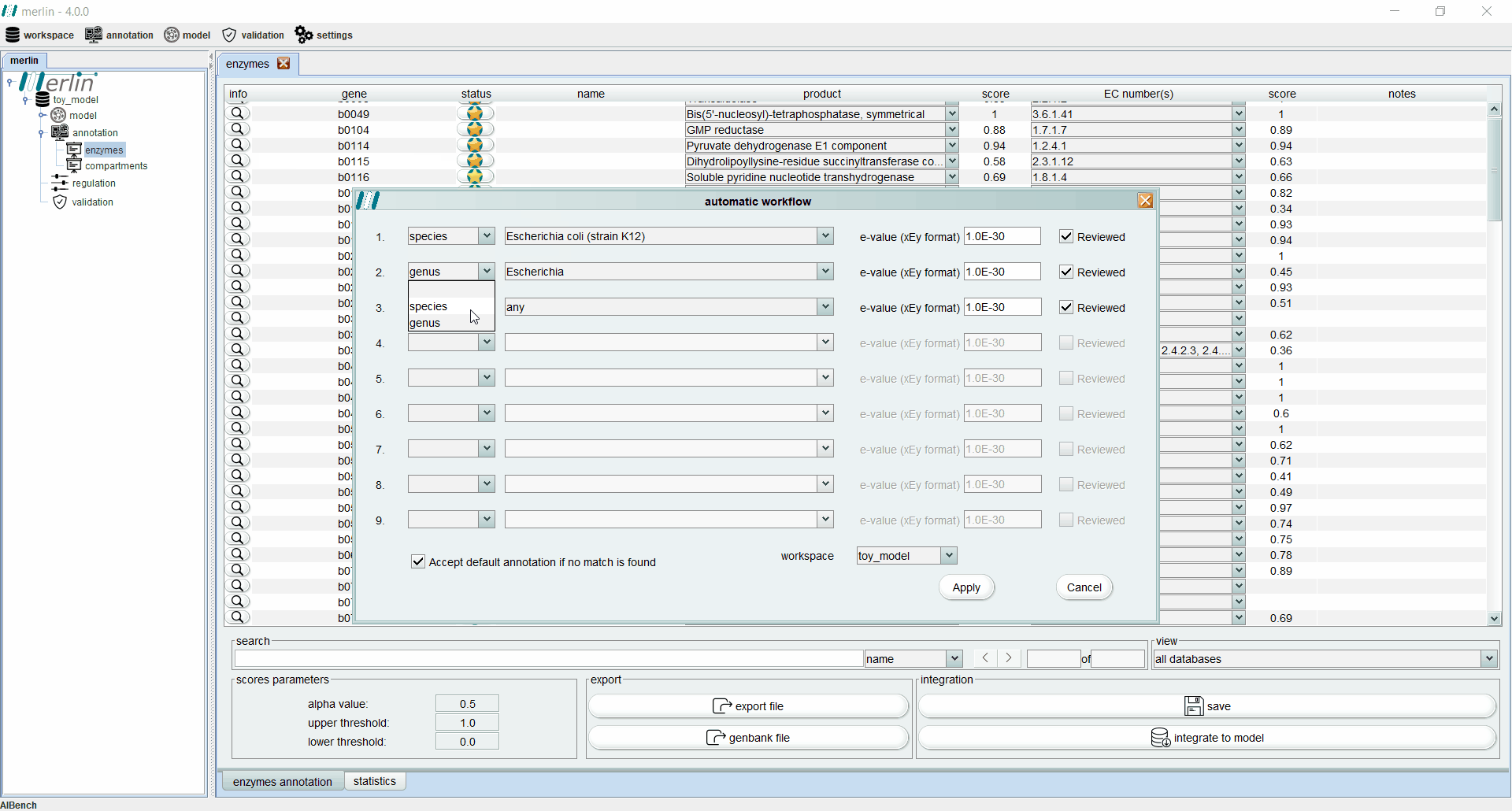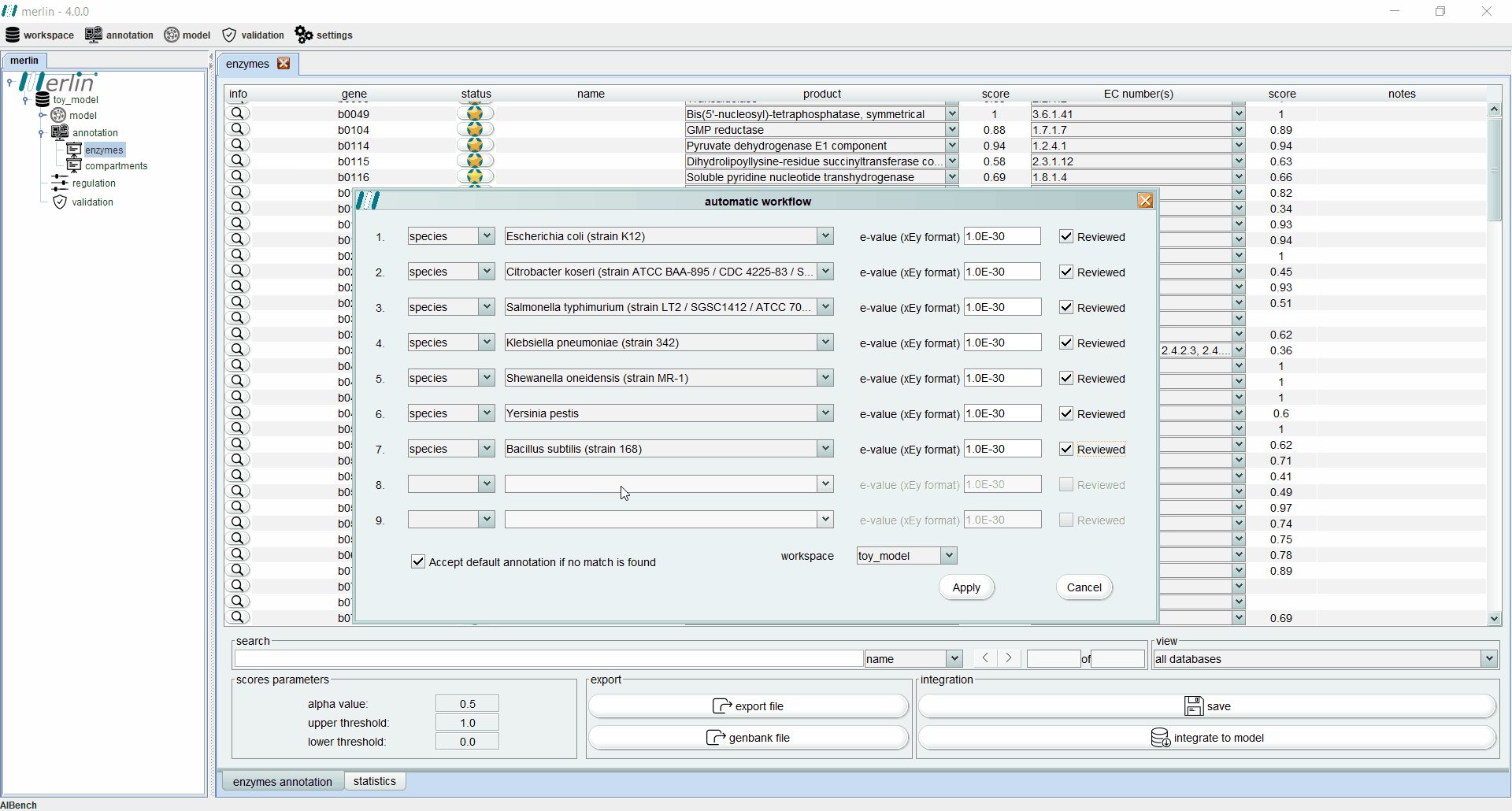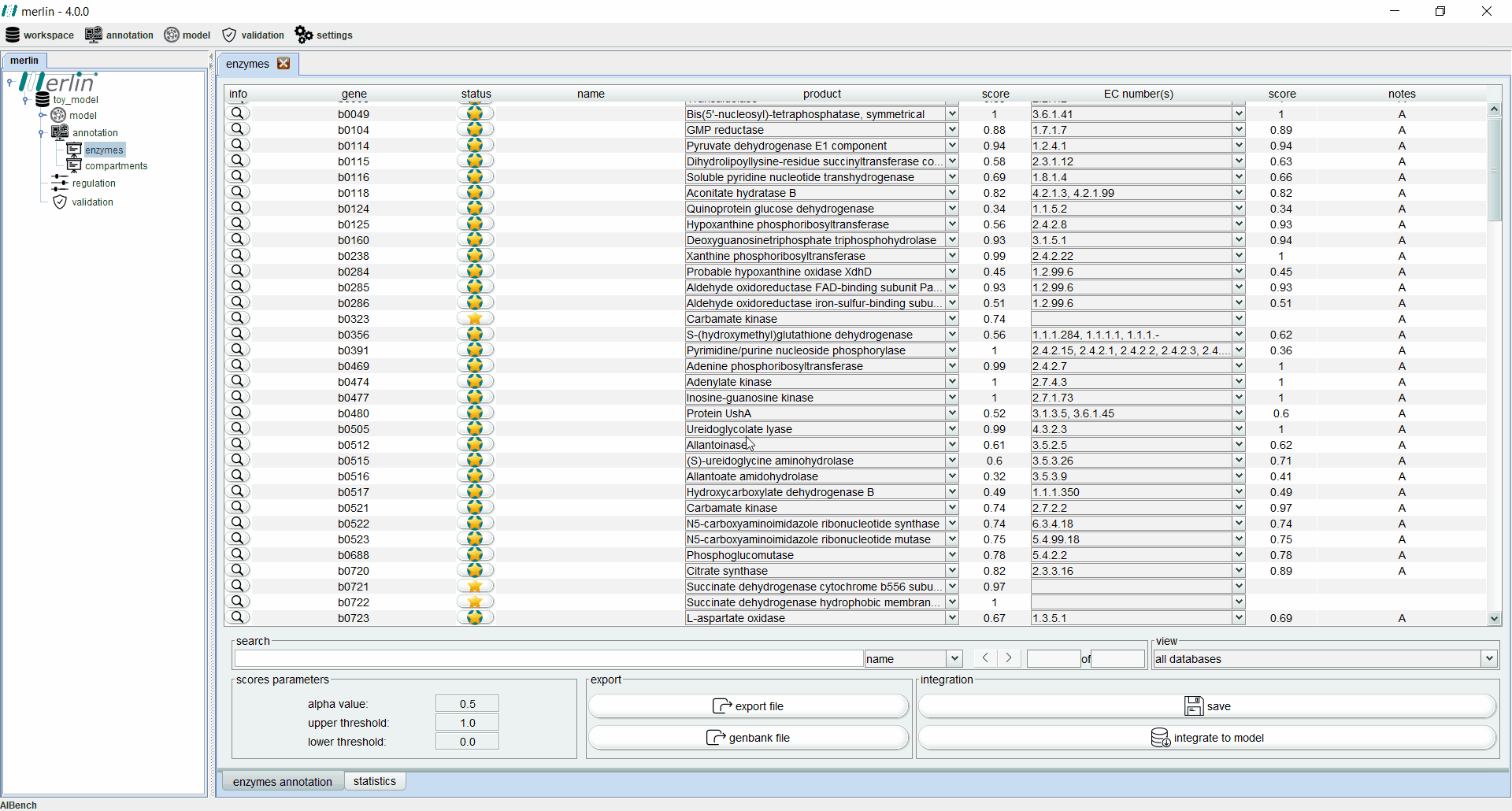
Enzymes annotation - Automatic workflow
This tool allows the user to iteratively replicate a simple annotation workflow. It consists of a GUI containing several slots (lines) with 4 fields (columns) to select parameters from the workflow. Such fields are:
a drop-down list in which the user can select between the species or the genus. If the empty option is selected, the remaining fields become inactive and excluded from the pipeline.
a drop-down list containing all species or all genus available in merlin’s internal database, depending on the option selected in the first field. Also the option “any” is available for both options, in order to accept any species or any genus.
the E-value threshold. Here, the user can define the maximum threshold above which all annotations must be disregarded.
field is a Boolean option in which the user can select between matches with UniProt reviewed entries or not.
In case that none of the conditions in each slot of the workflow is fulfilled, the user has available an extra Boolean field that allows the entries to accept the default annotation when no matches are found. The process of assessing the correct annotation, for each gene, starts by searching for the best match among the homologous genes found in the similarity search. With this purpose, an entry that meets the requirements of the first slot is sought among all available homologous genes. If no entries match the requirements, the process is repeated for the next slot, until the entire workflow is iterated. If no entries meet the requirements of any slot, the default annotation can be accepted or rejected, depending on the value of the Boolean option “Accept default annotation if no match is found”. A confidence level is associated with each annotation depending on which slot of the workflow the match was found. The confidence level is expressed in sequential alphabetical order. The default annotation also has a confidence level of “Default”. When the evaluation process is complete, merlin saves the annotation into its internal database and refreshes the Enzymes Annotation board to reveal the results of the process (see Enzymes board). The confidence level of each annotation is shown in the column “notes”.
SamPler
SamPler is a semi-automatic method for the selection of parameters for the annotation of enzymes. When merlin’s homology search is complete, a score is assigned to each EC number (and product name) by weighting the taxonomy score, frequency score through an α parameter. However, the best value for the parameter α is case oriented and has to be determined manually or using SamPler. To automatically determine the best value of α, the user should manually annotate a small sample of the genome (5% to 10%), allowing merlin to accept this sample as standard of truth.
SamPler is available as a plugin at “annotation/enzymes” menu. When this option is selected, merlin will launch a warning saying that all previous enzymes annotation will be deleted. At this point, the only options are to cancel the execution or continue. Note also that this tool can only be executed if BLAST already processed the whole genome. If the operation is continued, merlin will open a window in which the sample to annotate is provided. If required, a new sample can be generated using a different size. By pressing the button “save”, the current state of the annotation is saved and it can be continued in a different session. Thus, next time SamPler is executed, the same sample and manual annotations are loaded to the window.
During the annotation process, the tool provides a list of EC numbers that can be assigned to each gene. If the correct option is not available in the list, the option “other EC number” should be selected. Likewise, if a gene is not metabolic, the “empty” option should be selected. When the annotation of the sample is complete, the button “find best parameters” should be pressed. Here, SamPler starts a wide set of calculations that allow determining the best value of α, as well as an upper threshold and a lower threshold. When all calculations are complete, a results table is shown. This table allows analyzing the results attained for each α, namely of the accuracy, upper and lower thresholds, the number of genes above the upper threshold, number of genes below the lower threshold, total of genes for manual curation, the percentage of genes for manual curation and the precision/curation ratio. This window also allows to increase or decrease the values of the precision and negative predictive value.
The algorithm automatically selects the best parameters using the ratio as reference, the higher the ratio, the better the parameters. Nevertheless, the user may select a different option with different parameters based on the analysis of the results. By pressing the button “apply", the selected parameters are used for the enzymes annotation. Then, at the Enzymes Annotation board main tab, the scores are adjusted for the selected α parameter value, all entries above the upper threshold are accepted as correctly annotated, and below the lower threshold all entries are rejected as non-metabolic genes (see Enzymes board section). All genes between the upper and lower thresholds must then be manually curated. SamPler is published and more details are available at: SamPler a novel method for selecting parameters for gene functional annotation routines.
Enzymes annotation
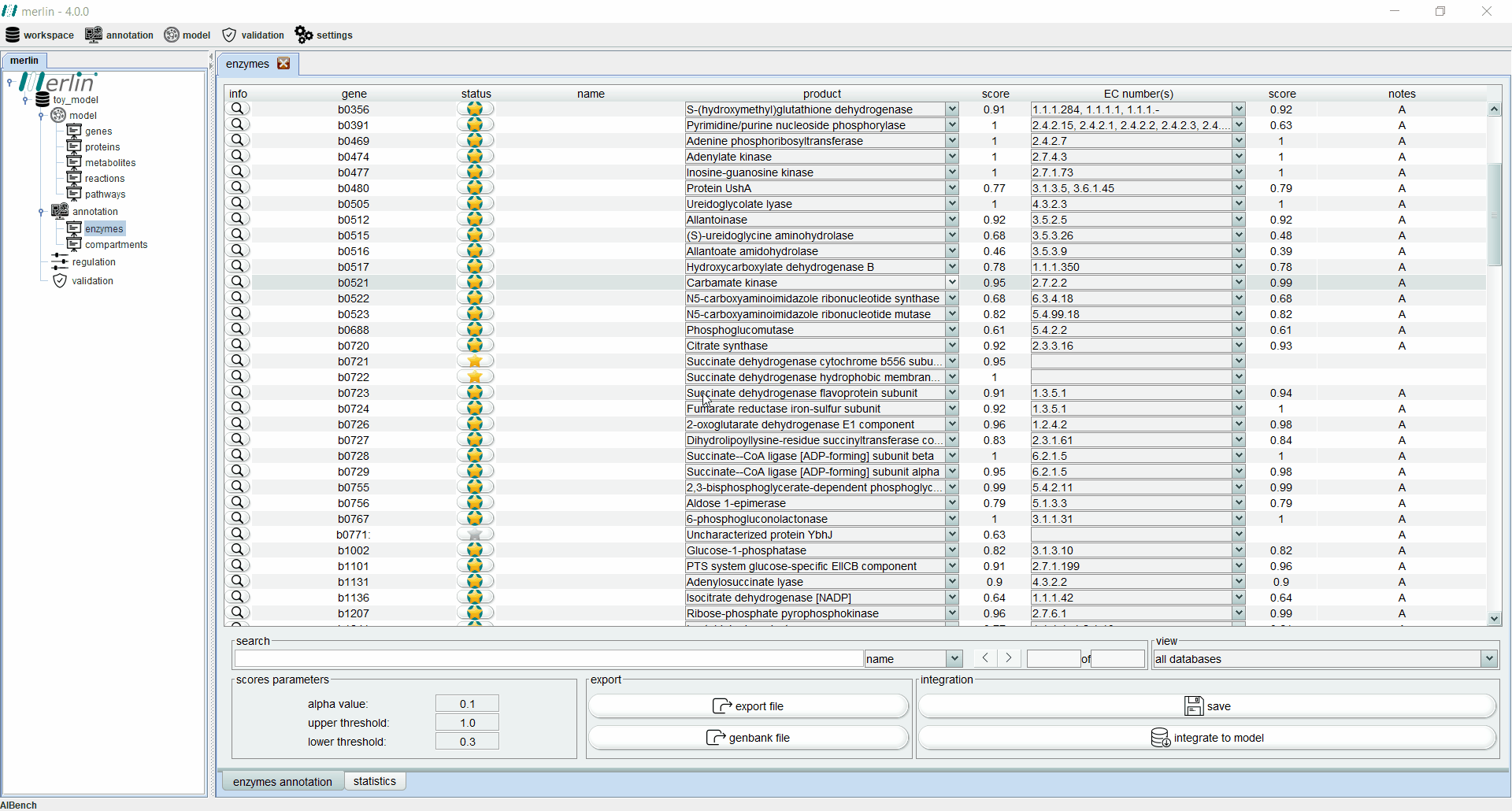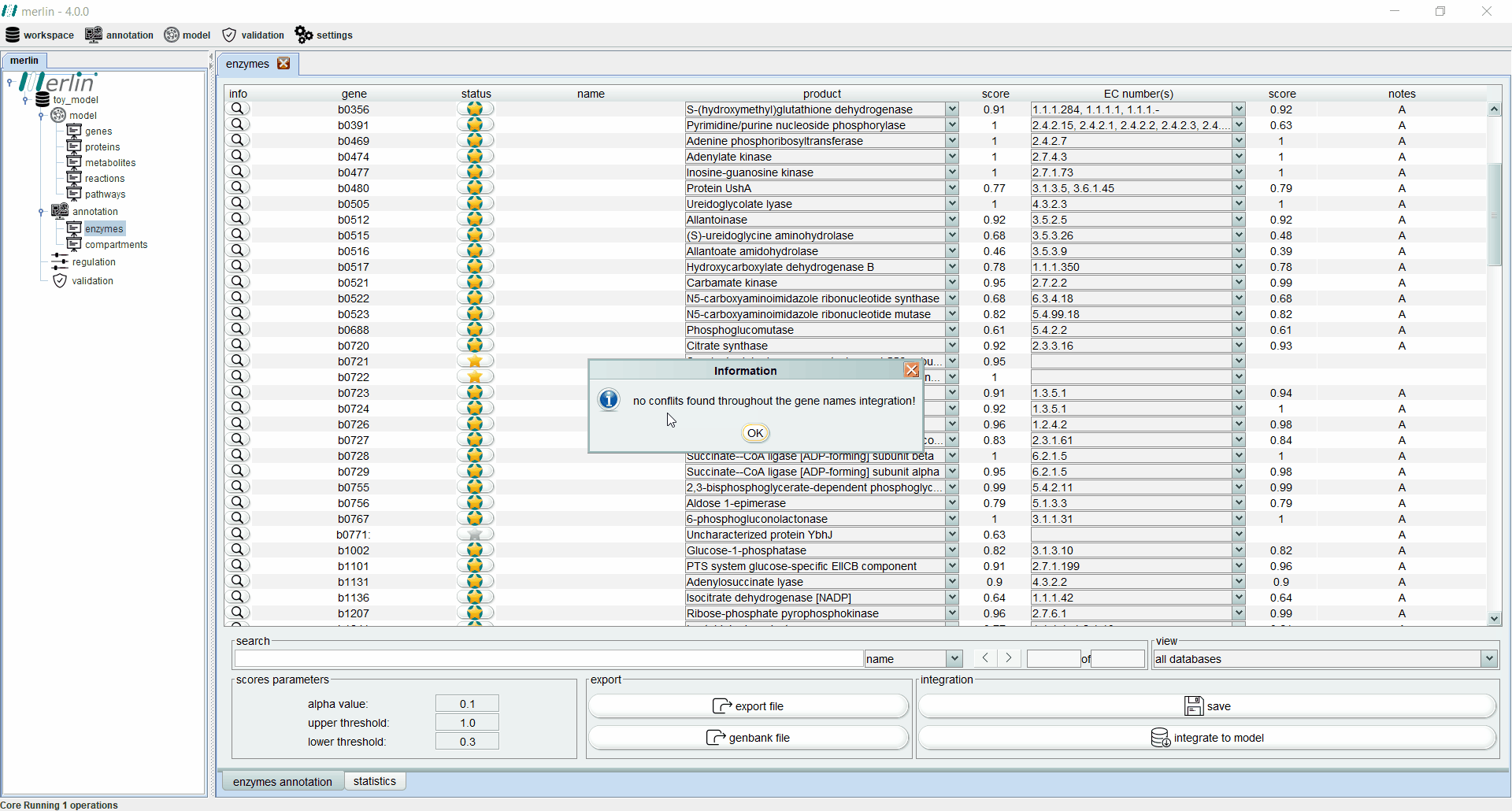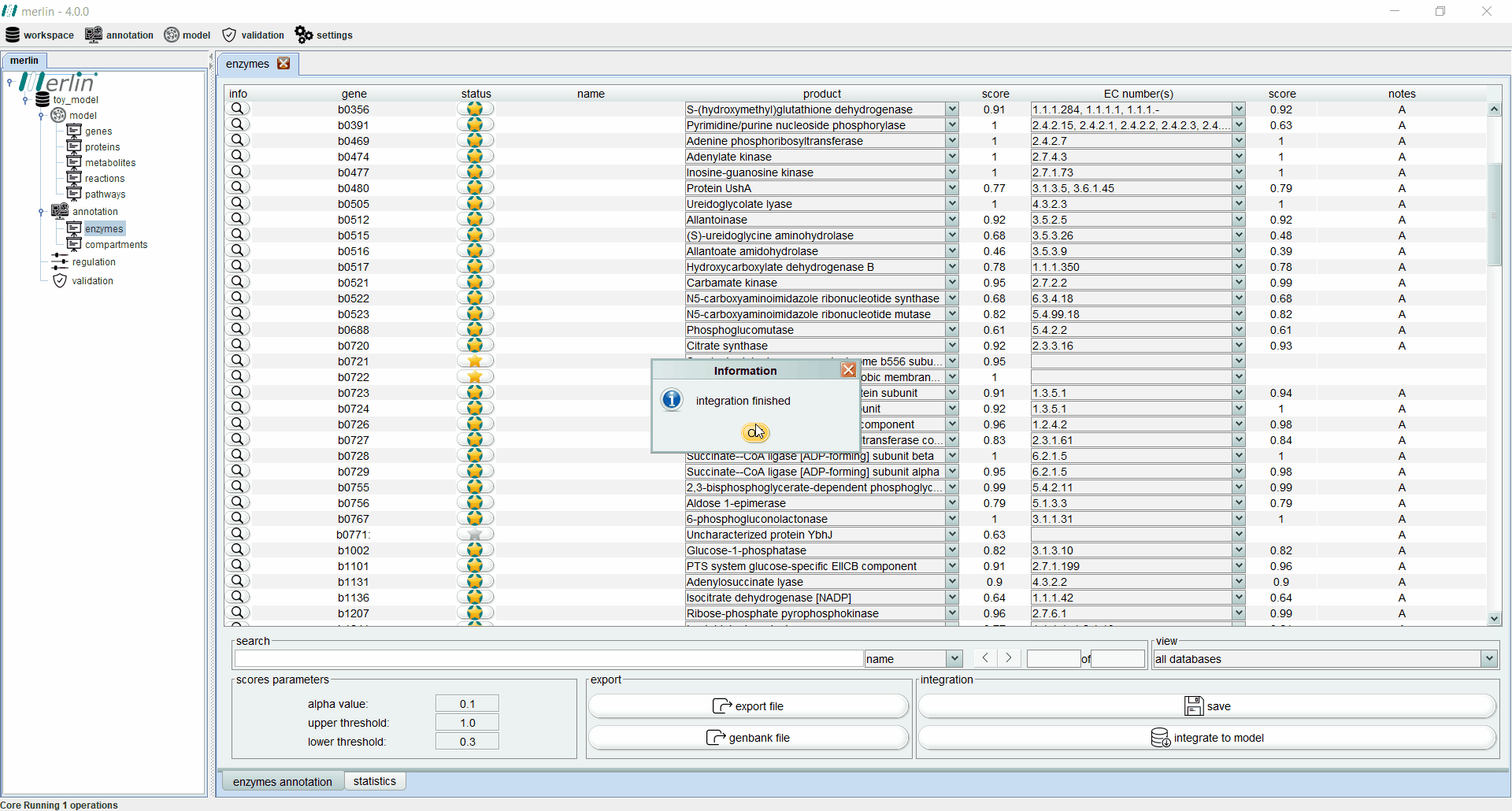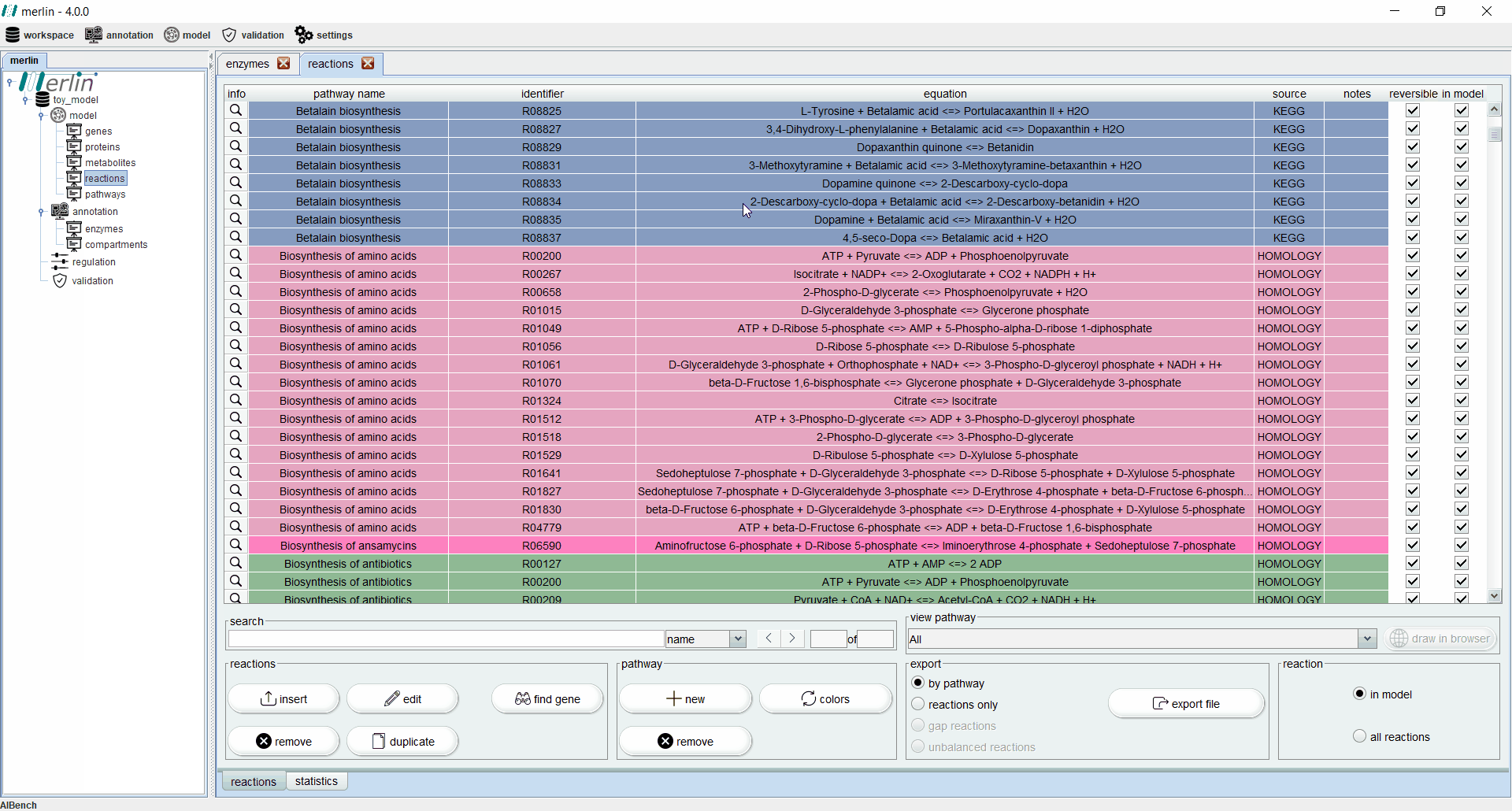
Enzymes board
The enzymes board shows, to the user, data related to the organism’s genome functional annotation. Its main tab presents all genes, as well as the associated status, name, product, EC number, scores and notes for each one. The status of a gene is divided in two components:
-
star: gene revision status:
-
Gold - Reviewed
-
Silver - Unreviewed
-
background: Match between merlin’s and UniProt's annotation:
-
Green - Match
-
Light Green - Partial match (it often happens with promiscuous enzymes)
-
Red - Mismatch
Additionally, each gene is assigned with a product and EC number based on score calculated by a merlin’s internal algorithm that takes into account both hits frequency and source organism taxonomy. However, products and EC numbers can be manually changed by the user, using the dropdown boxes available in each column. The enzyme board also contains a ‘notes’ column that allows the user to write notes for each gene (as a rule of thumb, the user should take note of any modification performed during the curation of the gene annotation, as backtracking changes in the model might be necessary). Additional information for each gene can be accessed using the magnifying glass, showing a panel with changeable tabs which show all hits obtained in the BLAST, protein sequence, BLAST parameters and organism taxonomy.
The search toolbar can be a valuable tool for the user, as it can search by gene name, by notes or in all columns. Setting the search toolbar with ‘all’ allows the user to search for EC numbers in all gene annotations, which can be useful in the later stages of the GSM model reconstruction. The main tab of the enzyme board can also be set to show only results from a selected database. Annotations shown in the board can be exported to a Microsoft Excel file or used to update the GenBank file of the organism’s genome. Finally, the two ‘buttons’ in the bottom right section of the board are used to save changes made in the board and to integrate the annotations to the model.
The board also displays a second tab that shows statistical information, such as the number of genes, number of genes without similarities, average number of homologues per gene, among others.

Enzymes annotation integration
merlin allows integrating the enzymes functional annotation into a draft genome-scale metabolic network very easily. The result of the integration between the loaded metabolic data and the enzymes functional annotation will define the topology and connectivity of your genome-scale metabolic network. The metabolic capabilities encoded in the genome previously identified with EC numbers will now be associated to reactions according to the mappings between enzymes (EC numbers) and reactions available in the metabolic data previously loaded into merlin.
The following example elucidates what is happening behind the scenes: A given gene has been annotated with the pyruvate formate-lyase EC number (2.3.1.54) in the enzymes annotation step; KEGG metabolic data has been loaded in merlin (see Metabolic data section); When this annotation is integrated into the model dashboard, the following metabolic reactions R00212 and R06987 are automatically associated with the referred enzyme and assumed to be present in your genome-scale metabolic model according to the enzymes functional annotation. merlin will perform this operation for all metabolic capabilities (genes assigned with at least one EC number) annotated in the enzymes functional annotation step and according to the loaded metabolic data. More details at: Reconstructing genome-scale metabolic models with merlin
In the Enzymes board, one can perform the integration of the enzymes functional annotation in the model dashboard using the Integrate to model button. This button will directly integrate the results of the enzymes annotation into the model dashboard and fill up the database tables assigned for this module.
Note that every time the Integrate to model button is used the results of the previous integration are lost. This operation saves the new results of the integration on top of the previous ones. Thus, although this button can be used to update the previous integration results, all alterations performed in the model dashboard and database tables designed for this module will be lost.
The Integrate to model button does not perform the Save operation in the enzymes annotation section. In other words, the enzymes functional annotation can only be saved in the database tables assigned for this purpose using the Save button in the Enzymes board.
Transporters annotation
TranSyT
merlin allows performing and integrating the transporters’ annotation into the draft genome-scale metabolic network, using the state-of-the-art tool Transport Systems Tracker (TranSyT). The TranSyT is a new approach to the problem of identifying genome-wide transmembrane transport systems, annotating these with reactions. TranSyT is the next iteration of TRIAGE (available in merlin v3), though much more efficient and designed to overcome its limitations. This new approach still relies on the TCDB to perform the annotation of transporters systems; however, TranSyT automatically retrieves and processes information from this source.
Run TranSyT
TranSyT is available as a plugin in merlin. By executing this operation, merlin will submit a request to TranSyT’s webserver, to generate transport reactions for the organism in study. When the results are retrieved, merlin will automatically integrate those reactions in the model and, if the compartmentalization of the model was already processed, assign compartments to the transport reactions. This operation allows setting “advanced options” for previously compartmentalized models. The fields internal/external/membrane compartment are set as “auto” by default. This means that merlin will try to find the compartments that better suit these localizations. However, if none are found, default compartments will be created for the ones missing. Alternatively, the user can type the name of the compartment to assign to each category, and if these compartments are present in the model, the operation proceeds.
When the process is complete, all reactions generated can be found in the model’s reaction board main tab at “Transporters pathway”.

Clean transport reactions
Cleaning transport reactions generated by TranSyT can be performed by selecting the operation available at: model - remove - transport reactions. This action will remove all TranSyT’s reactions from the model, which will no longer be available in the reactions board.
Compartments annotation
Compartments tools
merlin allows performing and integrating the compartments’ annotation into a draft genome-scale metabolic network using the results provided by Psortb3, WoLFPSORT and LocTree3. To load the reports generated by the aforementioned tools, access the “annotation” section, hover over the “compartments” option and select the “load reports” option. A panel will emerge asking for the respective workspace for the compartment report to be integrated into, and the tool used to generate the report. After clicking “proceed”, it is necessary to enter the URL with the compartment prediction report generated by the aforementioned tools available at the following platforms:
-
Psortb3 - It is worth noting that if a user wishes to integrate a report generated by Psortb3, besides the URL with the results it is also possible to submit a local file with the results’ output format as “Long Format (tab delimited)”

Compartments board
After uploading the reports of either one of the compartments predictors supported by merlin, all predictions should be present in this board. Here, results like primary compartment predicted, secondary compartments predicted and respective scores are available. The field “secondary compartments”, containing a drop down box with several different values and set as 0.1 by default, allows the visualization of the secondary compartments with a score similar to primary compartment score (difference of less than the selected value). The compartments shown in this table are the ones that will be considered for integration into the model.
Compartments integration
In the bottom right corner of the compartments annotation board main tab, it is possible to find the button “integrate to model”. This allows to assign compartments to the reactions in the model. This step is accomplished by checking the enzymes encoded by each gene and the reactions catalyzed by such enzymes. Thus, the reactions associated with such enzymes, will be assigned with the compartments annotated for the encoding genes in the compartments annotation board. By pressing this button, a new window, containing a table with all compartments predicted to be related with the genes submitted for prediction and an optional feature that allows the user to assign the default compartment membrane is shown. In this table, the user can select which compartments should be ignored by the compartmentalization operation. This feature is useful in cases in which some of the predicted compartments do not make sense in the organism in study. For instance, for cases in which the predicted compartment is “unknown”, or “chloroplast” for bacteria.
Regarding the option to set a default membrane (default membrane when compartmentalizing transport reactions), merlin allows the user to set this property manually or use “auto” mode. In “auto” mode, merlin will automatically search for the compartment related with the plasma membrane. If no compartment is found, merlin will launch a warning informing that the search was unsuccessful, and will automatically create a default compartment named “plasma membrane”. When the integration process is complete, the reactions in the model reaction board main tab should be compartmentalized. Note that the column “source” at the reactions board has now switched to “localization”.

Clean compartments integration
By selecting the button “clean integration” available in the annotation compartments board, all compartments assigned to the reactions in the model will be removed. This will cause the reactions shown in the model reactions board to revert to the non-compartmentalized state.
Biomass formulation
e-Biomass equation tool
merlin allows creating a biomass reaction for the draft genome-scale metabolic network. The formulation of the biomass reaction is one of the most important steps in your reconstruction. This is also the first step towards the conversion of our genome-scale metabolic network into a model. The biomass reaction should account for all the macromolecules that make up the biomass of the organism of interest.
merlin provides the e-biomass equation tool, which allows creating automatically the biomass reaction. This reaction includes DNA, RNA, Protein, Lipid and Carbohydrate macromolecules, as well as the group of cofactors. These metabolites will be denoted as electronic metabolites, namely e-Metabolites. The contents of each e-metabolite in the biomass reaction will be taken from the selected template. Alternatively, these contents can be manually assigned by the user. The table below shows the biomass templates available in merlin and the respective contents for each macromolecule.
Template |
Protein (g/gDW) |
DNA (g/gDW) |
RNA (g/gDW) |
mRNA (g/gRNA) |
tRNA (g/gRNA) |
rRNA (g/gRNA) |
|---|---|---|---|---|---|---|
Archaea |
0.61 |
0.034 |
0.227 |
0.05 |
0.15 |
0.80 |
Cyanobacteria |
0.595 |
0.016 |
0.077 |
0.05 |
0.15 |
0.80 |
Gram-positive |
0.588 |
0.016 |
0.077 |
0.05 |
0.15 |
0.80 |
Gram-negative |
0.533 |
0.027 |
0.136 |
0.05 |
0.15 |
0.80 |
Mold |
0.367 |
0.006 |
0.040 |
0.05 |
0.15 |
0.80 |
Yeast |
0.420 |
0.026 |
0.066 |
0.05 |
0.15 |
0.80 |
Besides the biomass reaction, the e-biomass equation tool will create a reaction for the main macromolecules whose composition might be estimated from the genome sequencing contents (DNA, RNA and Protein) (doi.org/10.1515/jib-2016-285) and for a group of previously determined Universal cofactors (doi:10.1016/j.ymben.2016.12.002) that make up the biomass of the organism of interest. For the former group, merlin uses the following data to perform the estimation:
-
gene translated amino acid sequences, i.e genome protein fasta file ( .faa), for the protein assembly
-
mRNA, i.e. genomic coding sequences fasta file (.fna) as well as tRNA and rRNA, i.e. RNA sequences fasta file (.fna), sequences for the RNA assembly
-
gene nucleotide sequences, i.e. genome sequence fasta file ( .fna), for the DNA assembly
Additionally, gene expression data can be used to adjust the amino acids contents. All cofactors in the e-Cofactor reaction are assigned with the same content. Note that, all reactions can be manually edited in the Reactions board (see below the Reactions board section). Furthermore, new reactions can be created for the assembly of other macromolecules in the Reactions board. For instance, the growth-associated energy requirements characterized by the number of ATP molecules required for synthesizing one gram of biomass can be added to the biomass as the hydrolysis of the energy currency of life.
Creating the e-Biomass equation
The user can select one of the biomass templates available in merlin. Alternatively, the user can customize the fraction of each macromolecule in the biomass reaction, by accessing the tools configurations sub-menu available in settings and selecting the e-biomass contents option. To create the biomass equation the user should select the e-biomass equation tool available in the model sub-menu called create. To use the manually set contents select the custom template, otherwise the most adequate template should be selected

e-Biomass equation composition
merlin can automatically estimate the amino acid, deoxyribonucleotide and ribonucleotide contents for the average protein, DNA and RNA, respectively. For that, the user must have manually or automatically retrieved and uploaded all genome files, namely the protein fasta file (.faa), the genomic coding sequences fasta file (.fna), the RNA sequences fasta file (.fna) and the genome sequence fasta file (.fna). See Genome files section for more detail. Leave the calculate protein, DNA and RNA checkboxes ticked for merlin to determine these contents automatically.
e-Biomass equation settings
The e-biomass equation tool allows selecting the source of the metabolic data used to determine the contents of each amino acid, deoxyribonucleotide and ribonucleotide. Currently, merlin supports KEGG and ModelSEED metabolic data. A biomass compartment can be created specifically for the biomass reactions in the biomass compartment field. Otherwise, leave this field as default for assigning the biomass reactions to the cytoplasmic compartment. In the advanced options, the user can use gene expression data to adjust each amino acid content in the protein reaction by providing the file path and data separator.

e-Biomass equation results
The results of the e-biomass equation tool will be displayed in a specific pathway called Biomass pathway in the Reactions board (see Reactions board section).
Drains reactions
Creating drains reactions
Exchange reactions or drains are a set of reactions necessary to replicate environmental conditions in which organisms survive and grow, as these can control the metabolite uptake and excretion. merlin provides a tool that automatically creates an exchange reaction for each metabolite in a given compartment. The user may manually insert the compartment for drain creation, if intended, or leave merlin’s default compartment, which is defined as ‘auto’. If the latter is chosen, the software will automatically search for an ‘extracellular’ or ‘outside’ compartment in the model and generate drain reactions for all metabolites in the compartment.
All drains generated by merlin are grouped into a pathway named ‘Drains Pathway’. Additionally, these reactions contain a single compound as a reactant, no products and its lower and upper bounds are set at a value of 0 and 999999 (default), respectively. These restrictions limit the input and output fluxes of a metabolite and should be constrained according to the environmental condition. For instance, to represent a metabolite available in the media, the lower bound of its drain should be restrained to a negative value. Fluxes determined in experiments may be used to constrain these reactions as well.

Cleaning drains reactions
As GSM model reconstruction is an iterative process, metabolites available in the model may change throughout the reconstruction. Therefore, merlin contains a tool which allows the user to automatically clean all the drain reactions previously generated by the drain reactions tool.
GPR associations
Creating GPR associations
The gene-protein-rules are generated using boolean logic in order to assemble GPR relationships of different complexities. merlin allows the user to create GPR associations for the draft genome-scale metabolic network using KEGG Orthology data. For each reaction in the model associated with one or more enzymes the algorithm looks for the KEGG orthologues associated with each enzyme. The genes associated with each ortholog are evaluated and a similarity search against the whole genome is performed with previously defined similarity thresholds. The similarity thresholds for both reference organism and other organism can be defined in the method ”gpr rules” inside “tools configuration” in the section “settings”. These values allow the algorithm to select the genes that should be selected after the similarity search.
The enzymes are connected to their orthologues through one or more modules which have a specific definition, comprising different orthologues, depending on the reaction they are associated with. The definition is used to generate the boolean rules using the similarity search results to accept or reject a definition. For example, if in a definition such as “(GeneA AND GeneB)” one of the genes is not selected in the BLAST evaluation procedure the definition is ignored. On the other hand if both genes are accepted the definition is accepted and can be used as one of the boolean rules for a reaction. The user may change these boolean rules in the “edit” button on the reactions view and apply the changes to the model.

Change GPR settings
merlin allows selecting many standards, such as “reference organism similarity threshold”, to create GPR associations. To assign different values to the thresholds you can access the GPR rules settings in “tools configuration” in section “settings”.
merlin boards
Boards for manual curation
merlin has been designed to foster the curation of draft genome-scale metabolic networks. For that, merlin provides diverse tools allowing to curate the model semi-automatically.
Genes
The “genes” board main tab lists all genes including its locus tags, names, number of encoding subunits, number of encoded proteins and number of associated reactions. Further information regarding a given gene is available when clicking the magnifying glass associated with each entry to access synonyms, orthologs, encoded proteins, associated compartments and sequences (DNA and Protein). Furthermore, this board contains multiple filters and operations below the gene table. The “search” field eases gene filtering by name or locus tag, highlighting the matched gene (the arrows and indexes on the rightmost position allow cycling between multiple matches). In addition, it is possible to filter the genes shown in the board by encoding genes only.
The “insert” option triggers a new window which allows users to create a new gene entry, while the “edit” option triggers an operation to modify existing genes. The “remove” option removes a given gene both from the board and from the database. The “export file” option triggers an operation to export listed data for each gene in the board in XLSX format. The “genes” board provides a statistics tab summarizing essential data, such as the total number of genes, number of protein encoding genes and number of enzyme encoding genes.

Proteins
The “proteins” board main tab lists all proteins including, names, identifiers, number of involved reactions and number of encoding genes. When the magnifying glass is clicked, it is possible to visualize additional information such as the encoded reactions, encoding genes, Gene-Protein-Reaction rules (if available), associated pathways, protein synonyms and associated compartments. The search field allows users to filter data by protein name or identifier.
The “insert” and “edit” operations allow creating new proteins and editing existing proteins, respectively. The “remove” operation removes a protein both from the board and the database. In addition, It is possible to filter proteins by the ones available “in model” proteins, which are proteins encoded in the genome. Moreover, this board contains an additional statistics tab describing data, such as the number of proteins, number of enzymes and number of transporter proteins in the database.

Metabolites
The “metabolites” board main tab lists all metabolites information, namely: names, associated compartments, formulae, external identifiers, number of associated biochemical and transport reactions in the model. By clicking on the magnifying glass associated with a given metabolite, it is possible to visualize additional information such as reactions in which it is involved , synonyms and entry type. The search field allows users to seek metabolites by name, formula and external identifier.
The “insert”, “edit” and “remove” operations are responsible for creating new, editing existing and removing metabolites, respectively. Additionally, it is possible to filter the “metabolites” board by reactants, highlighting the entries in green, and by products, highlighting the entries in blue. All other entries are simultaneously reactants and products. As the “proteins” board , it is possible to filter the “metabolites” board by metabolites integrated in the model. This board contains an additional statistics tab describing data, such as the number of reactants, number of products, number of reactants which are both reactants and products and number number of associated reactions.

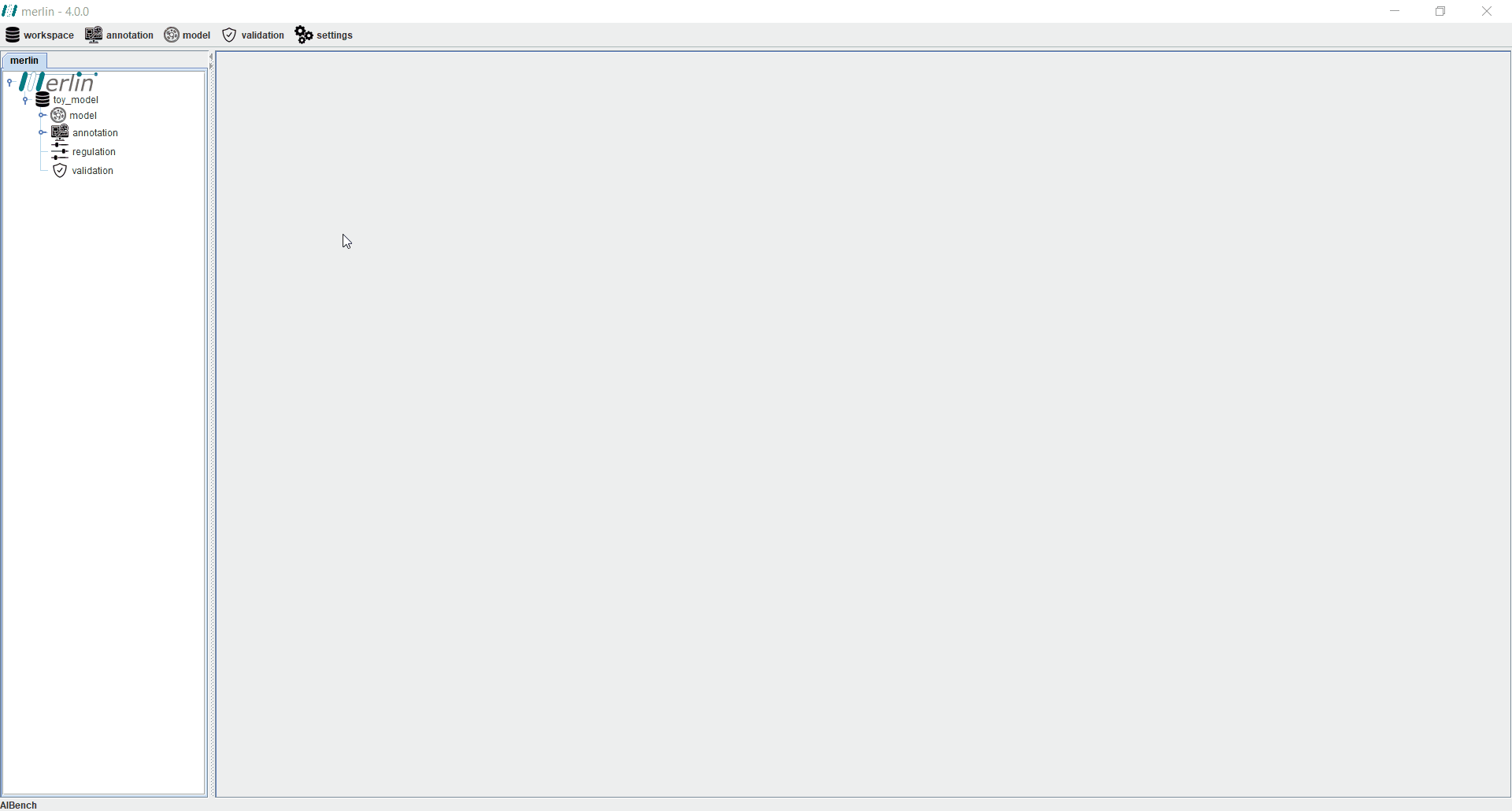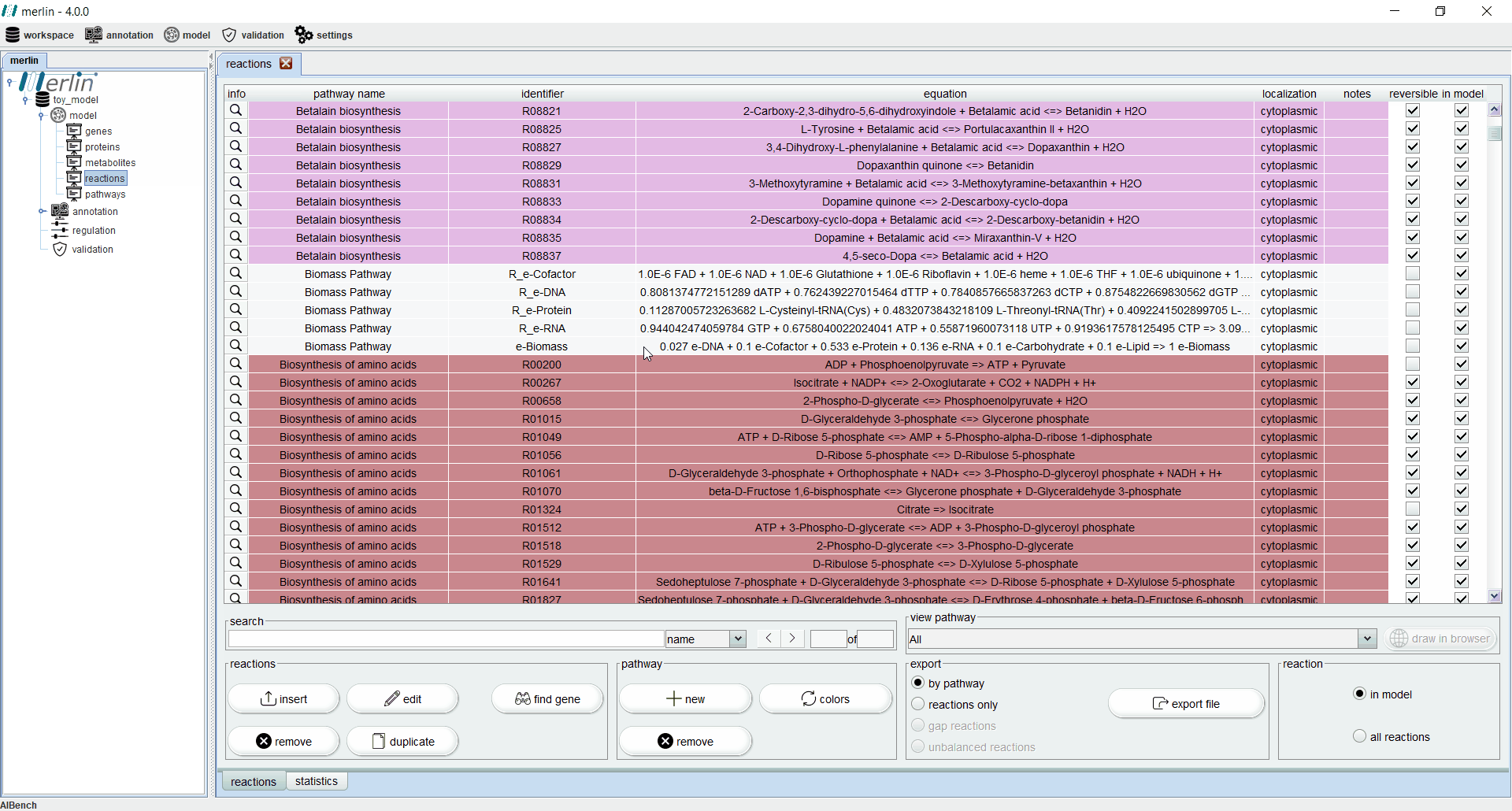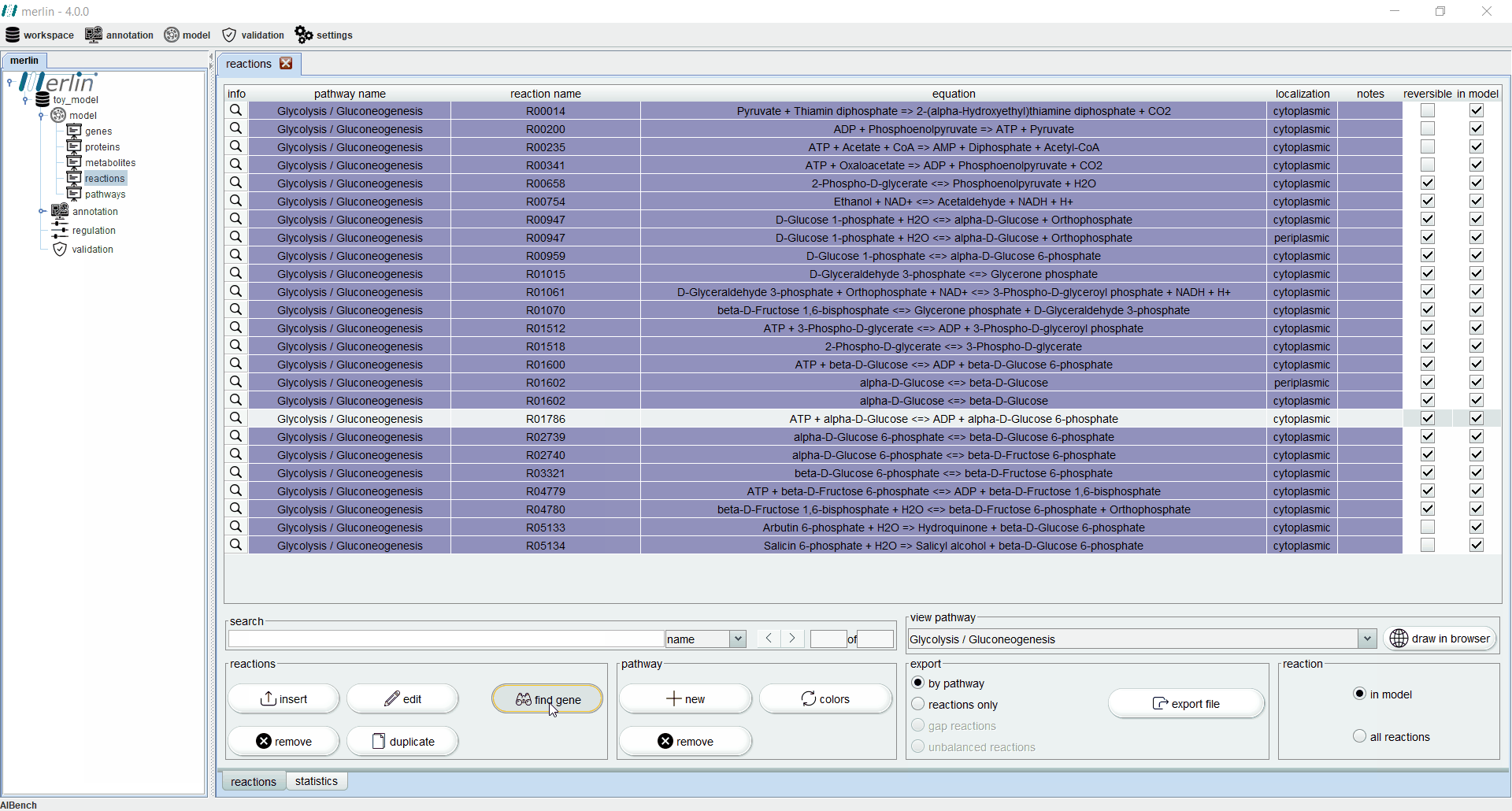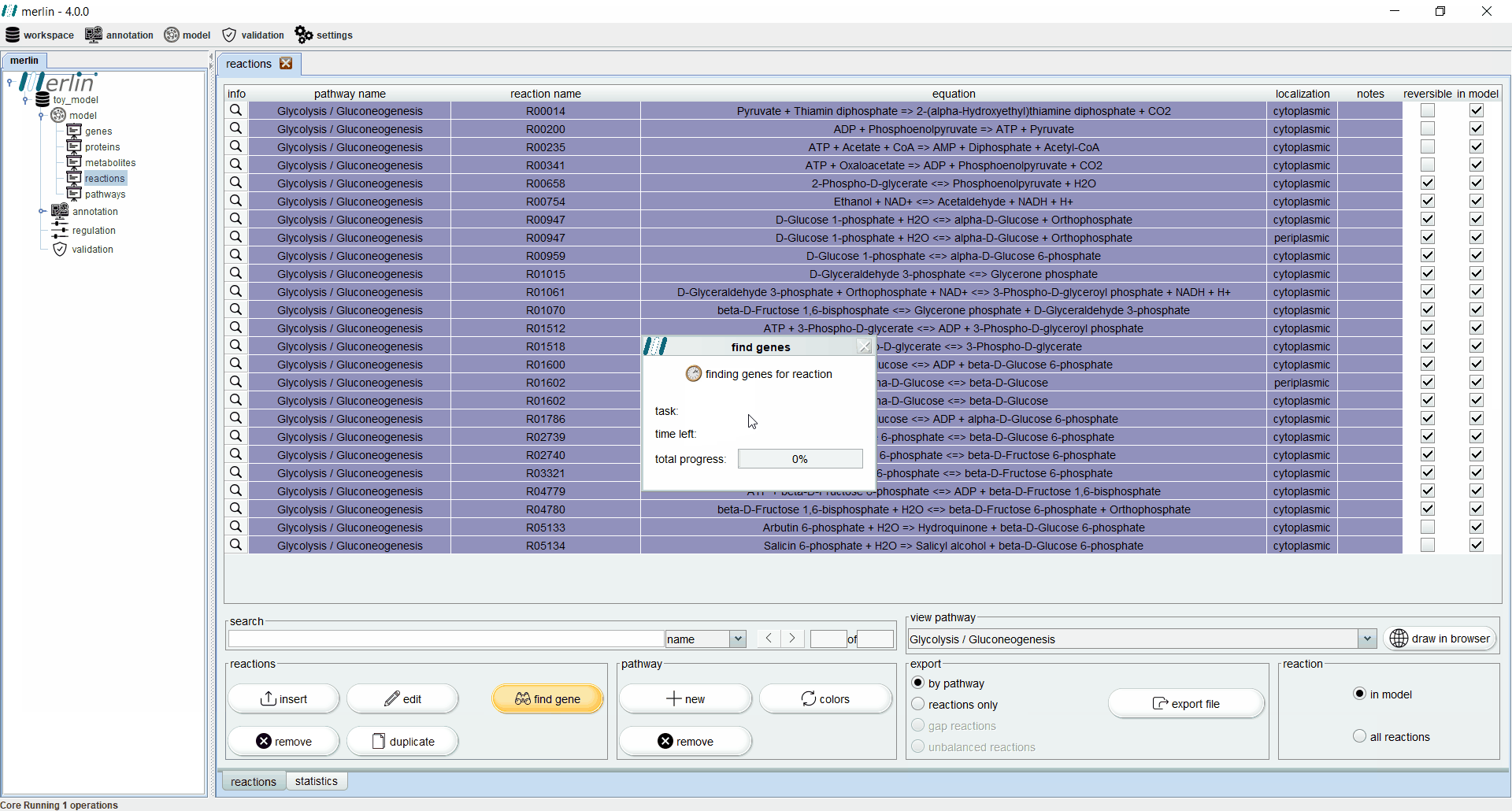
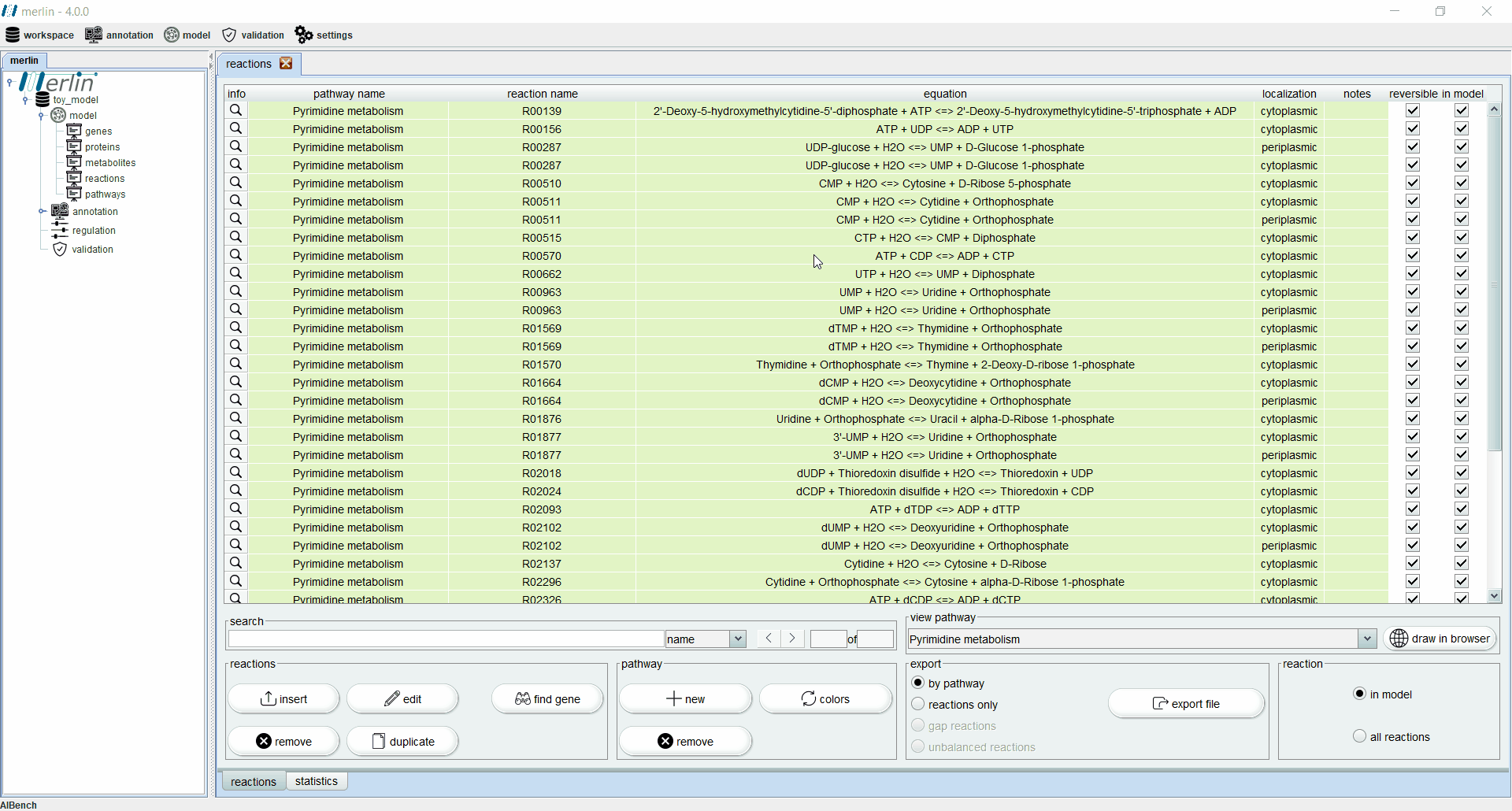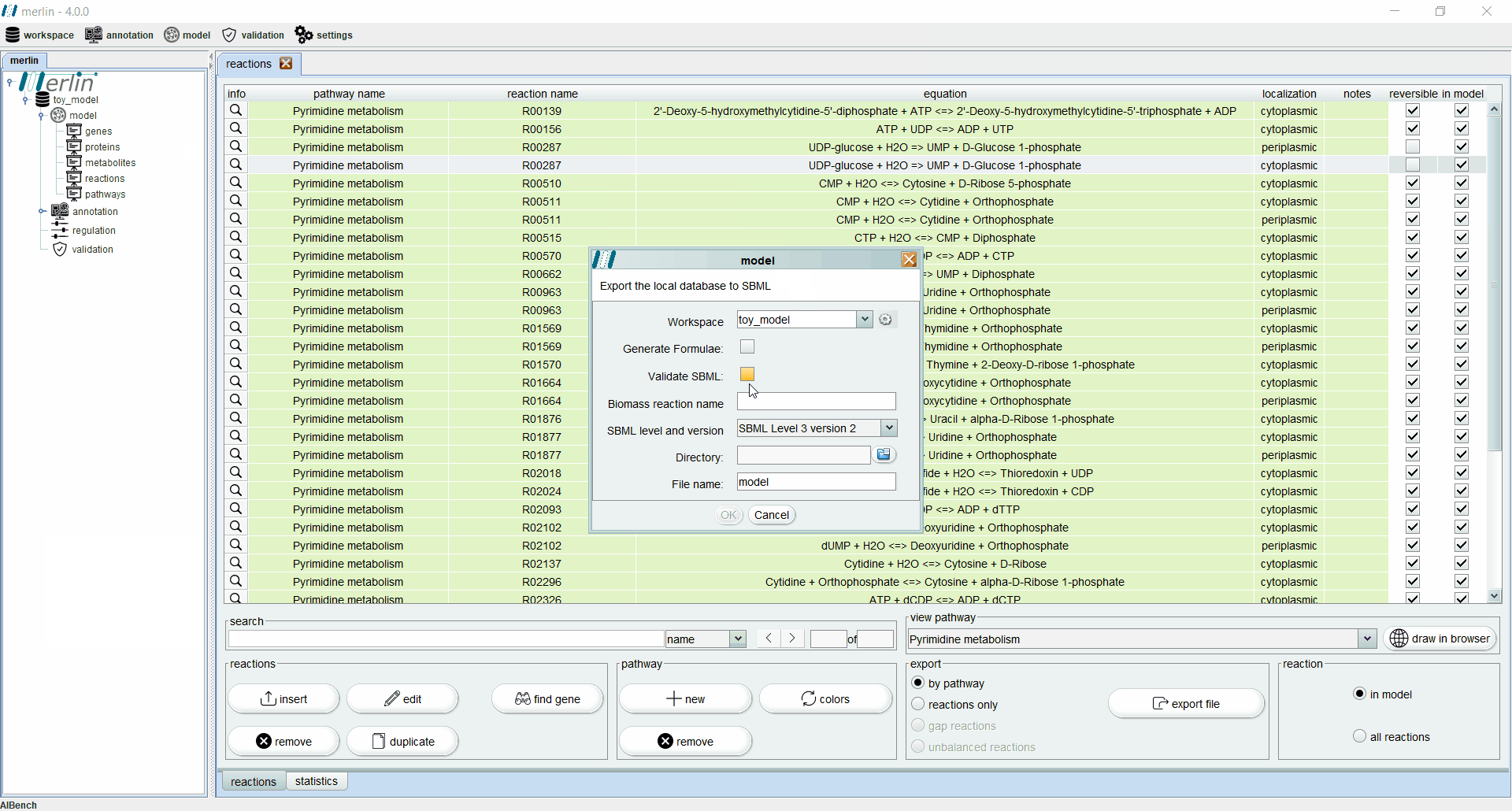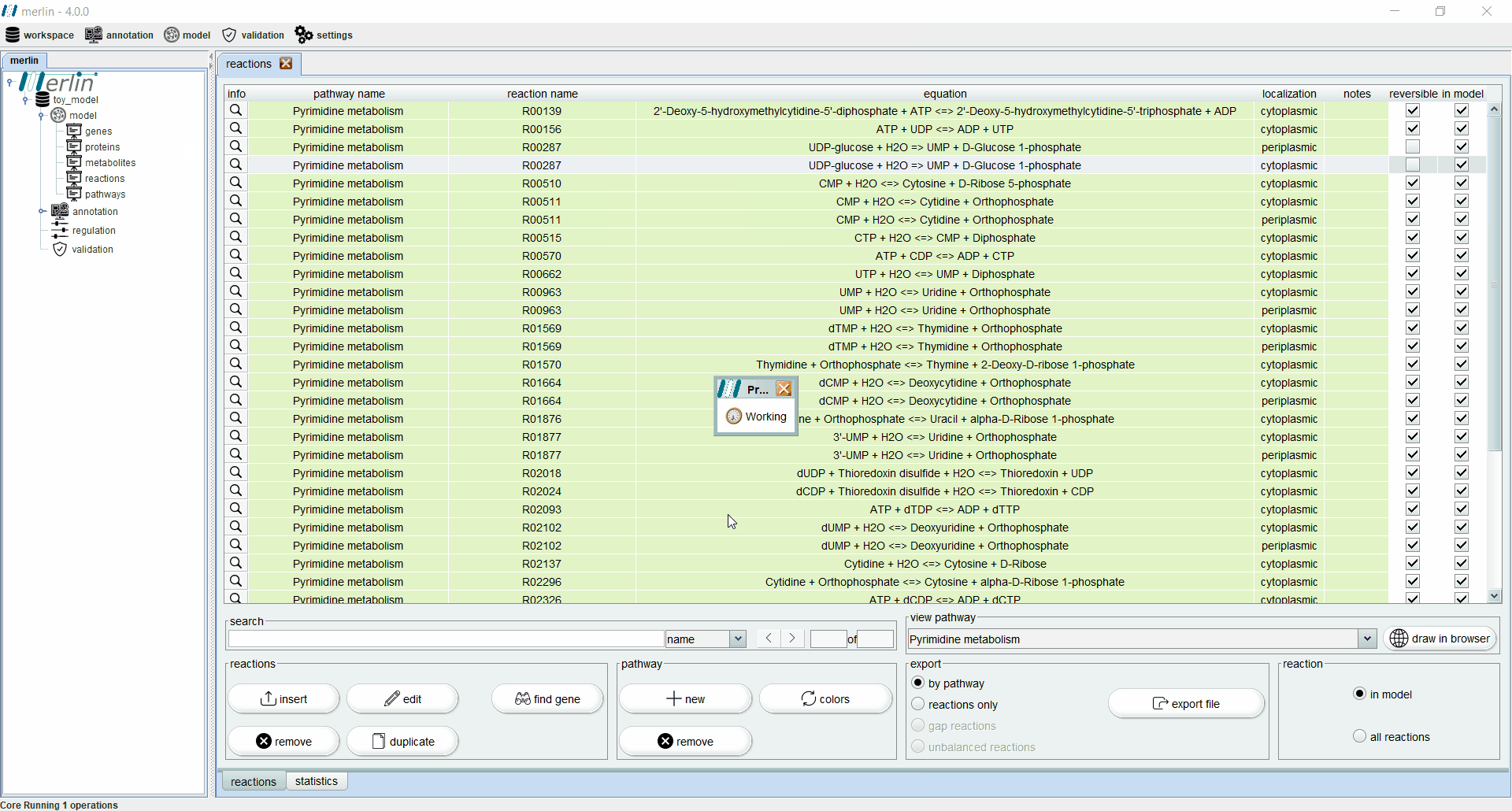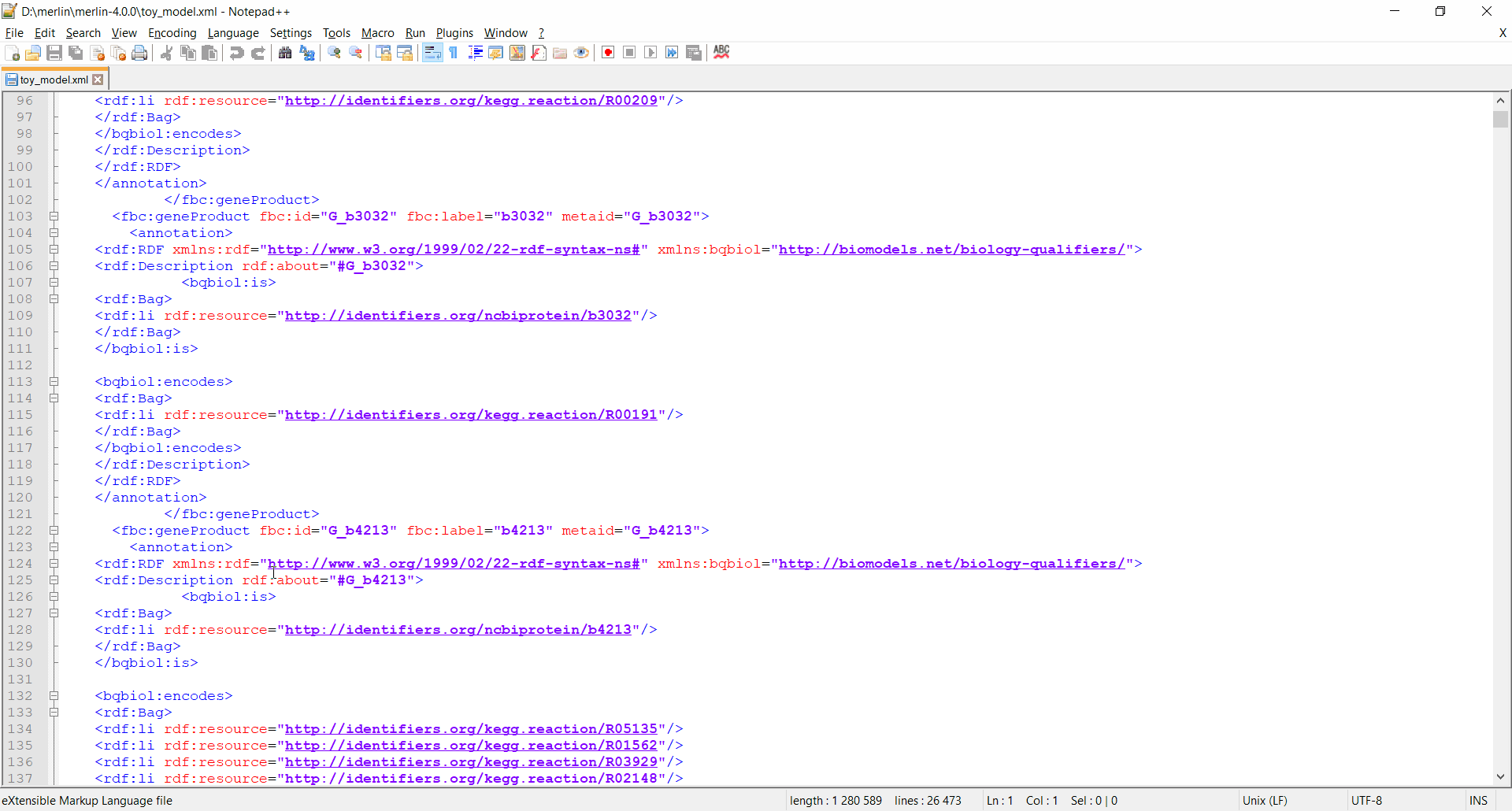
Reactions
The “reactions” board main tab lists all reactions including the equations, reaction identifiers, associated pathways and sources. The “source” column is an important feature for:
-
identifying manually created/edited reactions as “MANUAL”
-
reactions obtained from homology search results as “HOMOLOGY”
-
unprocessed reactions retrieved from KEGG as “KEGG”
-
reactions from TranSyT as “Transport”
-
reactions from other models as “SBML”
Additionally, each reaction is identified as reversible (based on upper and lower flux bounds) and as integrated in the model or not. The magnifying glass next to each reaction triggers a panel with additional data such the metabolites involved in the reaction, associated enzymes, reaction properties and synonyms, associated pathways and source. The search field allows users to filter reactions by reaction identifier and equation. Additionally, it is possible to filter the set of shown reactions by pathway. Furthermore, after selecting a specific pathway, by clicking in the “draw in browser” option it is possible to access an external webpage from KEGG, using the default browser, with a visual representation of the pathway as represented in merlin.
The “insert”, “edit” and “remove” operations are responsible for creating new reactions, modifying pre existing reactions, and removing a reaction respectively.
The “find gene” button triggers an operation to find an orthologous gene for a given reaction.
The “duplicate” button creates a copy of a chosen reaction, with the only change being the incremented reaction identifier (to distinguish copies from the original reaction). The pathway subsection contains the “new” and “remove” buttons, which trigger operations to create a new pathway or remove an existing one.
All reactions listed in the “reactions” board are color coded by pathway, thus every reaction belonging to the same pathway has the same color. The “colors” button automatically refreshes the color palette of the “reactions” board. The “export file” button generates a XLSX file with all reactions data, which can be further filtered by pathway.
Finally, it is possible to filter the “reactions” board to list only reactions integrated in the model. This board contains an additional statistics tab describing data, such as the number of reactions in the model filtered by source and number of reversible and irreversible reactions.

Tools
Correct reversibility
Incorrect reversibility and directionality of reactions in a GSM model can lead to futile cycles, which impairs model’s predictions. Additionally, all reactions retrieved from KEGG are reversible, thus its correction is mandatory. Hence, merlin offers a tool which corrects reactions (loaded in the database) directionality and reversibility using a database published by Stelzer et al (2011) or ModelSEED organisms’ templates as a source of corrections. This tool allows performing a forced correction, which is used when users have already performed this correction and want to reapply the correction.

Find unbalanced reactions
Unbalanced reactions in GSM models can lead to incorrect flux distributions during simulations. Hence, reactions’ stoichiometry has to be ensured, by guaranteeing that the atoms’ sum in reactions’ participating compounds is equal on both sides of the reaction equation. As in previous cases, merlin contains a tool that identifies unbalanced reactions. This operation requires a ‘proton name’ input (default: H+), which can be defined by the user. The tool sets the unbalanced reactions’ names in bold and italic to easily pinpoint unbalanced reactions. Additionally, for each reaction in the model, a new tab, named ‘balance’, becomes available in the dropdown box of the reaction data panel, accessed using the ‘magnifying glass’. Unbalanced reactions can also be exported in an excel file. The typical unbalanced reaction issues found within a metabolic network include:
-
metabolite missing formulae
-
missing/excess protons or water
-
macromolecule synthesis/breakdown
Usually, the first and second cases are easily solvable by determining the missing formulae and adding/removing protons/water, respectively, whereas the third situation requires the removal of the macromolecule on one side of the reaction equation. However, as no specific rules to correct these issues exist, one should always check biological databases such as MetaCyc and BRENDA, to obtain additional information, before correcting each unbalanced reaction.

Find blocked reactions
In some occasions, interveniens of reactions are neither consumed or produced by other reactions, which results in a ‘blocked’ reaction. In these situations, flux through the reaction during simulations is impaired, and, in some cases, could affect the model’s performance. Usually, blocked reactions are a result of errors in the enzyme annotation or compartmentalization of the model. merlin has an operation that aims at identifying these reactions, coloring them in red (blocked reaction) or light blue (leads to a block reaction) to aid the analysis of unconnected reactions. Additionally, in the reaction data panel, a new tab becomes available, stating which metabolites of the reaction are considered dead-end metabolites (metabolite that cannot be consumed nor produced). Therefore, the ‘find blocked reactions’ tool is a valuable asset to curate the GSM model, as it can help in the identification of metabolic gaps (missing reactions in a given pathway), correction of enzyme annotations and incorrect directionality/reversibility of reactions.

Remove blocked reactions
Usually, most unconnected reactions are unnecessary for a GSM model. Therefore, merlin offers the possibility to remove automatically all the detected blocked reactions using the ‘remove blocked reactions’ operation. However, before using said tool, it is mandatory to identify the blocked reactions firstly, otherwise the operation issues a warning.
Find gene
Ideally, all metabolic reactions in the model should be associated to a gene. The “Find Gene” tool identifies genes able to catalyse a given reaction. If the reaction is associated with enzymes in KEGG, this tool will seek genes, in the case study organism, similar to the KEGG knock-outs associated with such enzymes. The tool will filter all orthologous genes found, using previously defined thresholds. The tool will present a panel with the orthologous genes, together with information from the similarity search performed for each ortholog. The user can then select which genes should be associated with the reaction and included in the model.
BioISO
BioISO is aimed at evaluating your biomass formulation or genome-scale metabolic network. This tool is based on the Constraints-Based Reconstruction and Analysis (COBRA) and Flux Balance Analysis (FBA) frameworks. BioISO can receive as input the current genome-scale metabolic model and return potential errors in the biomass formulation or gaps in the metabolic network.
Run BioISO
To run BioISO, go to the validation menu and select a reaction to be tested and the objective direction (maximize or minimize the reaction). All reactions available in the model will be displayed in the dropdown box. It is expected BioISO to take a few minutes (2-5 minutes) depending on the model size, internet connection and server availability.
BioISO results
The results will be available under the validation dashboard on the left. One can open the results board identified by the submitted reaction. The results will be displayed in a tabular format by clicking on the corresponding board. The precursors (reactants) and products of the submitted reaction are displayed together with the analysis, number of associated reactions and role of the metabolite in the submitted reaction. The evaluation performed by BioISO for each metabolite is based on the analysis of the metabolic network capability to synthesize or consume that given metabolite, depending on the role. Then, you can take a look at the reactions associated with each metabolite by opening a new panel on the magnifying glass. In this panel, the evaluation result depends on whether the reaction can carry any flux towards the consumption or synthesis of the previous metabolite, depending on the role. The number of reactants and products is also available in this panel.
To get more inception about a given reaction, one can open a new panel by clicking on the associated magnifying glass. This panel will display BioISO’s analysis over the metabolites involved in that specific reaction. Finally, you can get more inception on the model state by repeating the last two operations over such metabolites and reactions
A pipeline for performing manual curation with BioISO
merlin easily allows you to curate your metabolic model. For that, a straightforward pipeline using BioISO is suggested. Once the user has assembled the draft genome-scale metabolic network, performed the reversibility correction, and resolved both unbalanced and blocked reactions, we suggest using BioISO to hasten the model curation and debugging.
To run BioISO an objective reaction must be provided. For instance, the main objective of microorganisms living in wild-type environmental conditions is to grow namely maximizing the biomass production. Following this principle, the first reaction to be submitted to BioISO might be the biomass reaction. BioISO will evaluate whether the current model is capable of synthesizing or consuming the metabolites associated with this reaction, thus attaining the biomass production. In addition to this preliminary analysis, BioISO will conduct its analysis to the reactions associated with these precursor metabolites and so on and so forth. In this way, one can solve potential errors in the draft genome-scale metabolic model by focussing specifically in modules that cannot carry any flux towards the objective. Note that such modules are just a given set of metabolites and reactions that cannot carry flux.
The manual curation process is therefore leveraged with this pipeline, as every reaction can be submitted as objective. This allows evaluating whether a given pathway can synthesize or consume a given metabolite,by analysing the reactions present in such via. The faulty modules (reactions or metabolites), according to BioISO results, should be thoroughly assessed. Finally, all potential errors in the model should be found and corrected them and BioISO can be used to reassess the same objective.
Export
Exporting a model
merlin allows exporting the model in SBML (level 2 or 3), a format based on XML used to represent computational models of biological phenomena. To export a model the user should access the “workspace” section, hover over the “export” option and select the “model” option. A new panel will emerge asking for the workspace which contains the model to be exported. Additionally, it is possible to export a text file containing the formulae of model metabolites by selecting the “Generate Formulae” option. After selecting the output directory and name, it is possible to export the model in SBML format.
Import
Importing a model
It is possible to import a pre existing model to merlin as long as it is available in SBML level 2 or 3 format. To perform this operation access the “workspace” section, hover over the “import” option and select the “model” option. A new panel will emerge asking to select the workspace which will host the model to be loaded. After selecting the correct SBML level of the model, a user only needs to select the actual model file.

Plugins manager
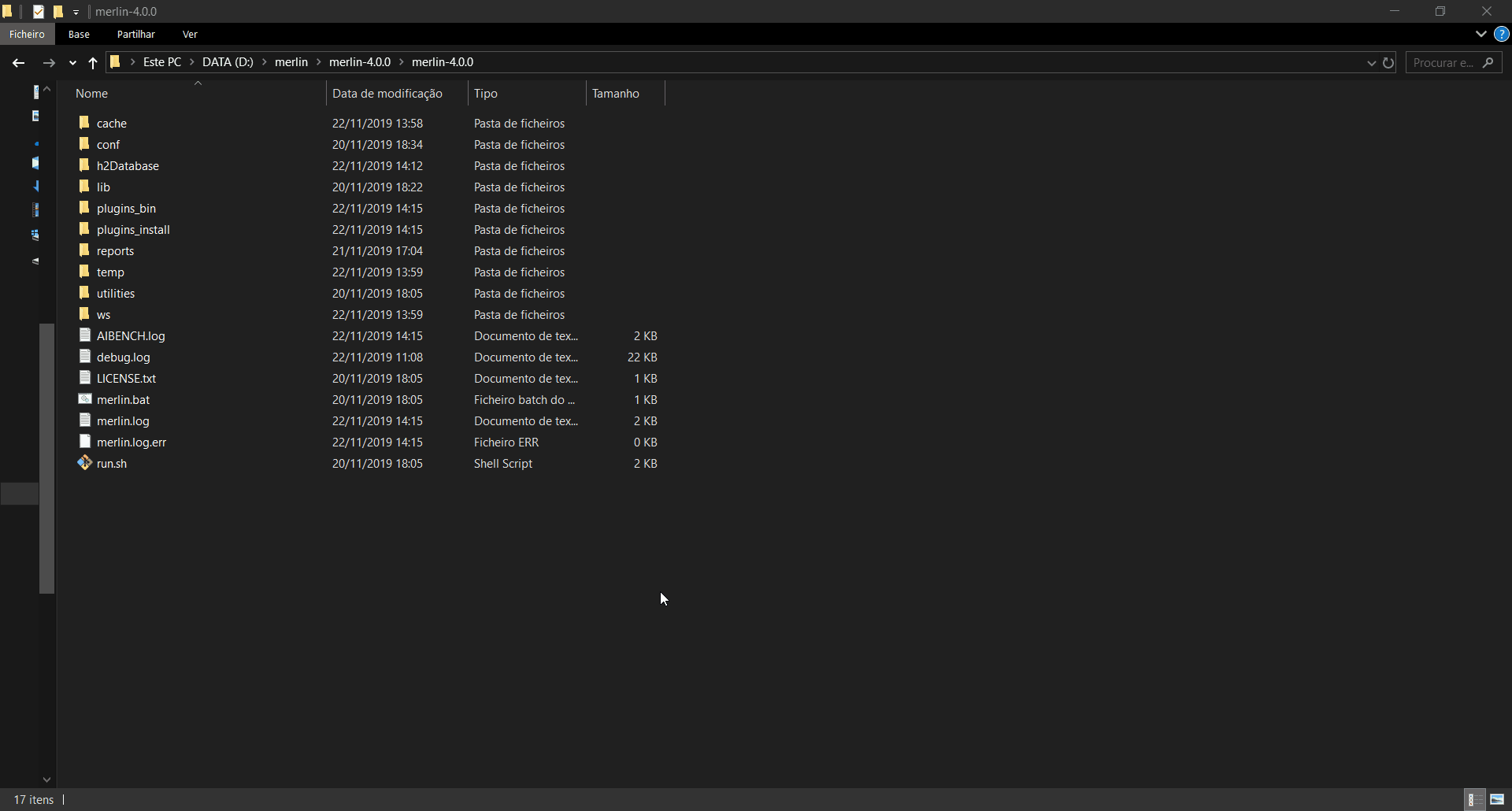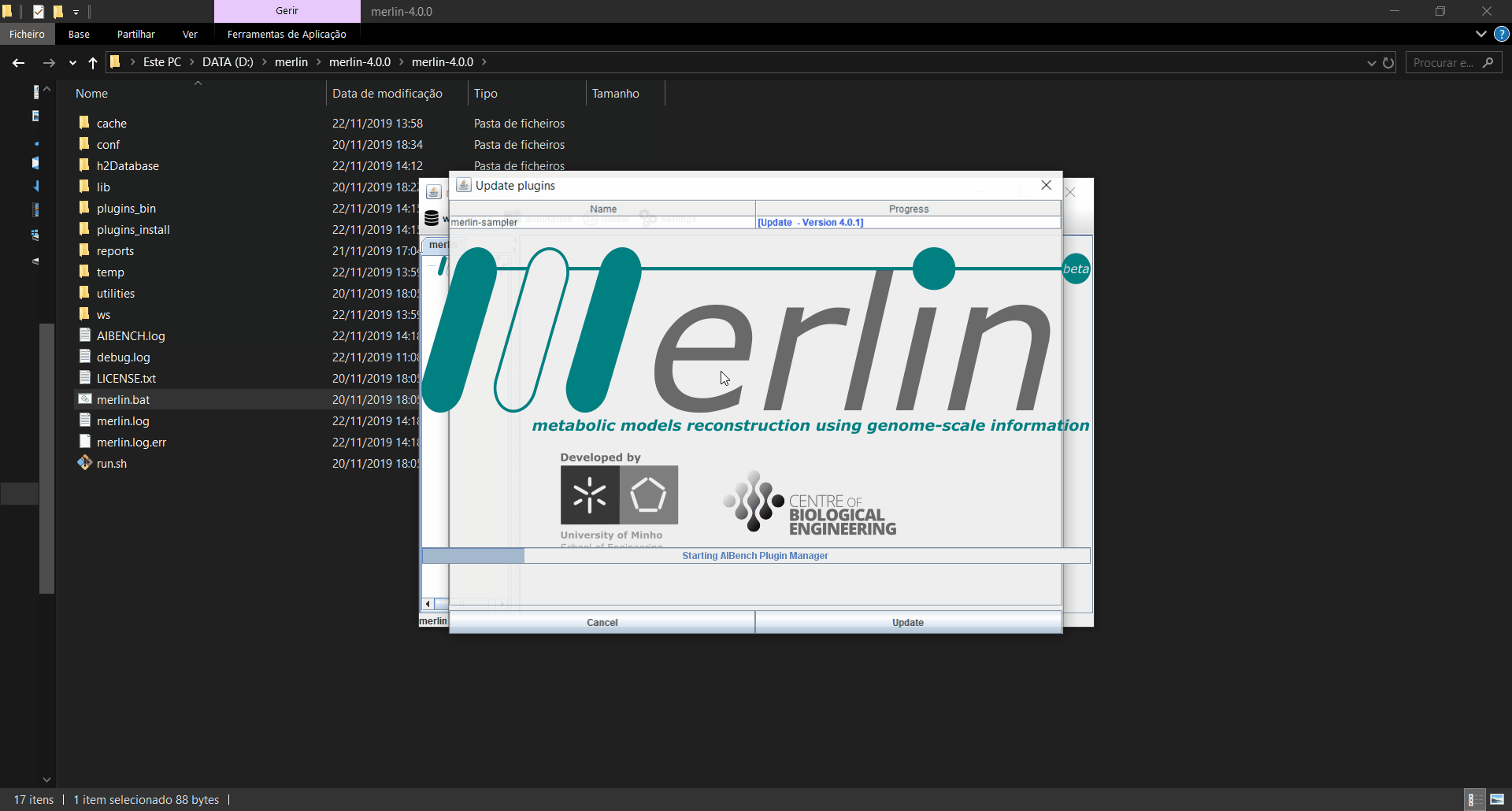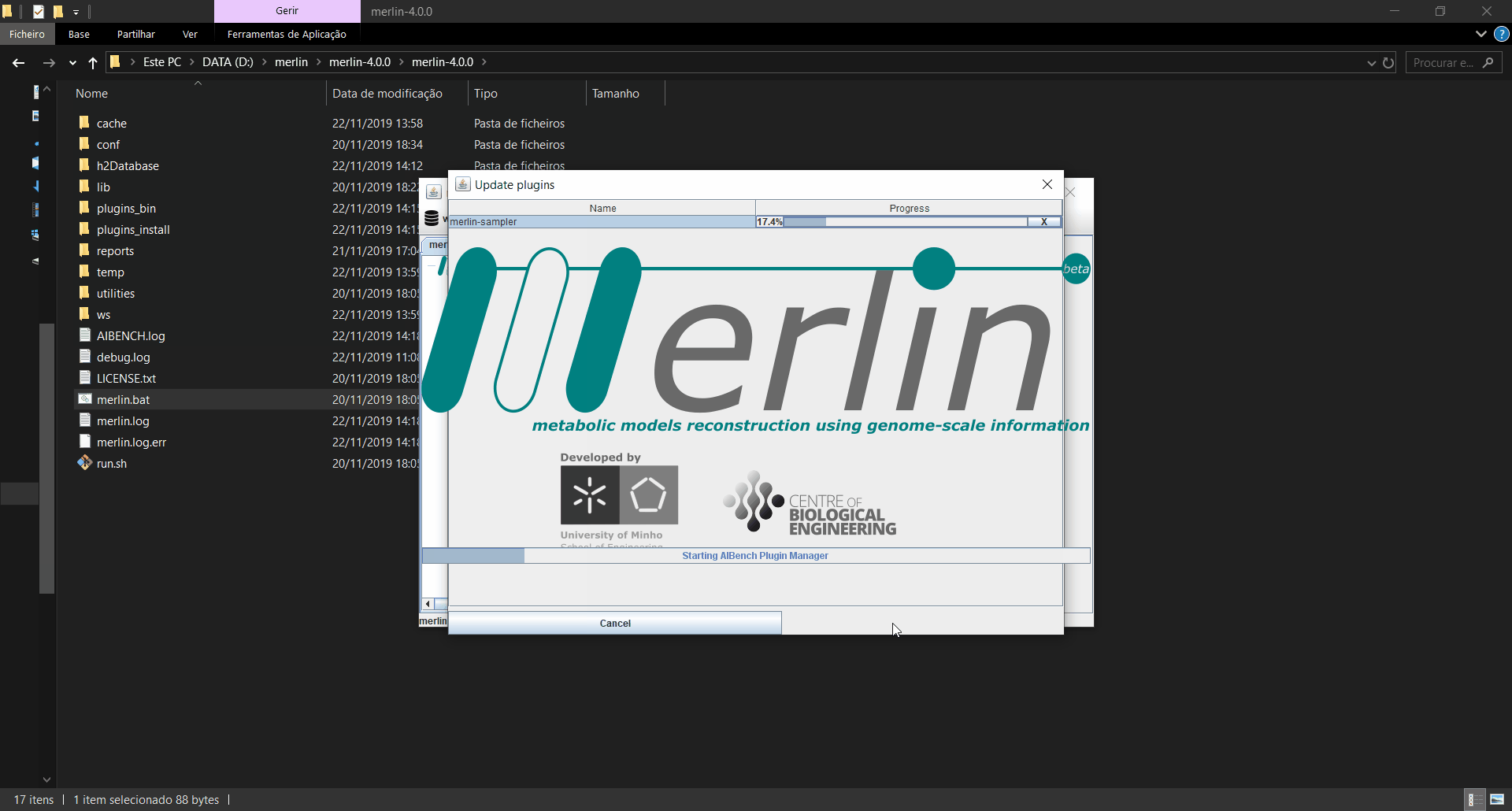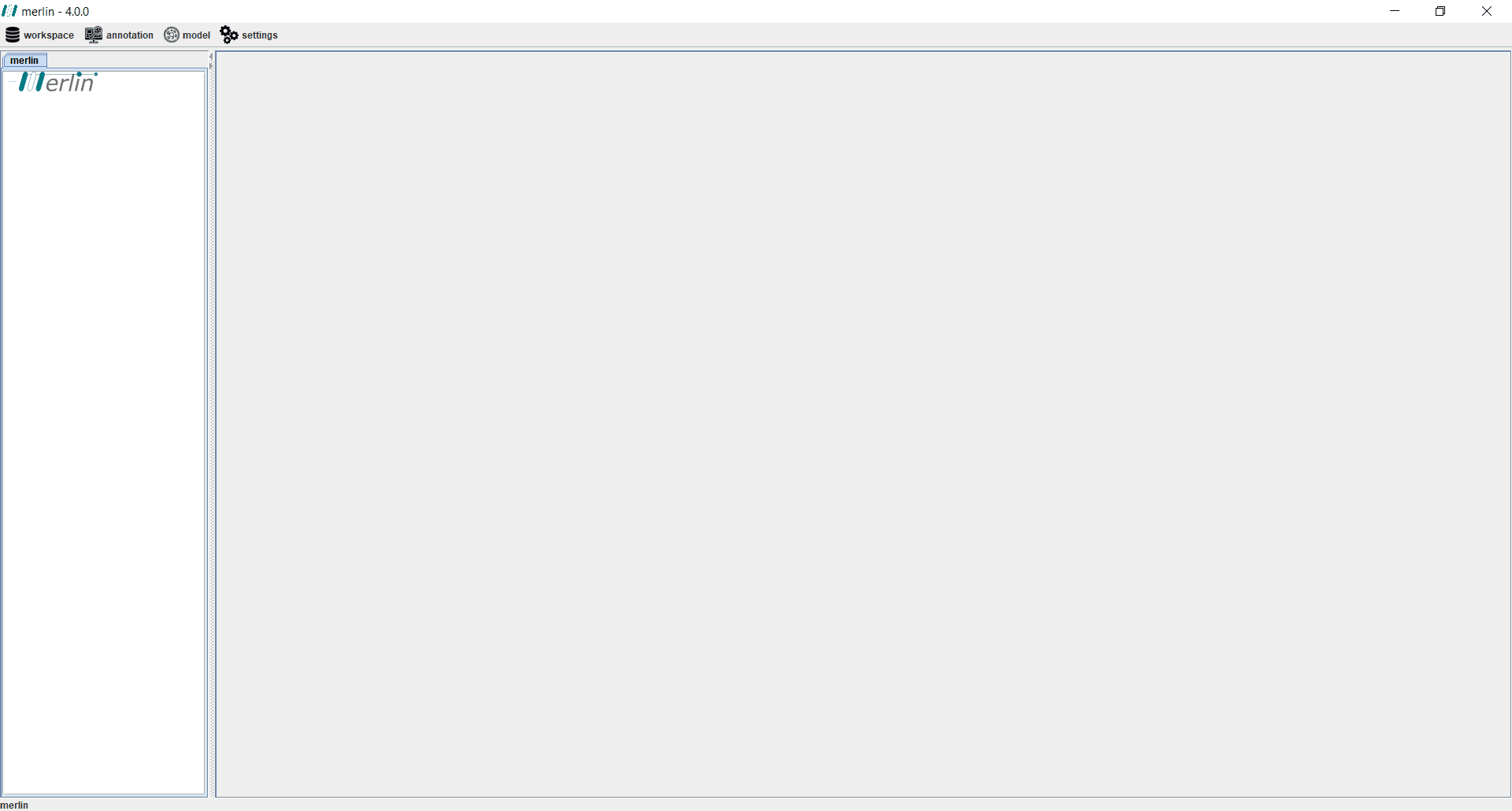
Updating plugins
When merlin is executed, it will automatically search for updates in the plugins installed, and if any updates are found, a window informing which plugins were updated is launched. If the updates are installed, merlin will restart for the changes to take effect. Also, an operation is available in the menu on the “settings” option to manually search for updates.
Installing plugins
The operation “repository manager” allows the user to enable, disable and/or install new plugins into merlin. The upper table inside the manager graphical interface allows to verify the plugins that are already installed in merlin as well as other information, and a checkbox to enable or disable each plugin. The lower table contains the plugins that are available in the online repository. To require the installation of a plugin, the button “Install - (...)” regarding the selected plugin must be pressed inside in the column “Download”. Any modification regarding the plugins in this board requires restarting merlin to take effect.

Other tools
Reset locus tag
After performing BLAST, genes’ identifiers might be altered and by integrating the enzymes annotation to the model, genes’ locus tags are changed. Therefore, this operation resets the identifiers to the original genes’ locus tags, using the local download genome files or by performing an online search.
Increase maximum heap memory size
It is recommend to increase the maximum heap memory size (RAM) for large genome sequences or multiple reconstructions. If the case, one can go to "settings" section in the menu and select "increase memory". While, the maximum heap memory size is set by default to 1GB, you can select a higher amount of maximum heap memory size in the dropdown menu.